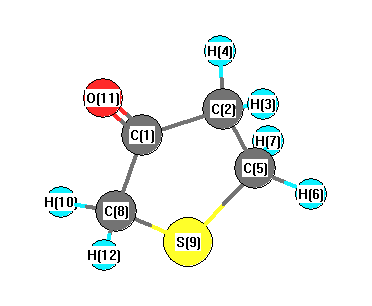
Vibrational Frequencies calculated at PBEPBE/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3067 |
3049 |
4.87 |
|
|
|
| 2 |
A |
3050 |
3033 |
0.82 |
|
|
|
| 3 |
A |
3048 |
3030 |
7.14 |
|
|
|
| 4 |
A |
2986 |
2969 |
27.40 |
|
|
|
| 5 |
A |
2972 |
2955 |
13.83 |
|
|
|
| 6 |
A |
2971 |
2954 |
4.64 |
|
|
|
| 7 |
A |
1773 |
1762 |
190.26 |
|
|
|
| 8 |
A |
1412 |
1403 |
1.96 |
|
|
|
| 9 |
A |
1356 |
1349 |
5.71 |
|
|
|
| 10 |
A |
1353 |
1345 |
12.72 |
|
|
|
| 11 |
A |
1240 |
1233 |
9.61 |
|
|
|
| 12 |
A |
1226 |
1219 |
20.17 |
|
|
|
| 13 |
A |
1171 |
1164 |
11.84 |
|
|
|
| 14 |
A |
1150 |
1143 |
30.37 |
|
|
|
| 15 |
A |
1108 |
1101 |
8.87 |
|
|
|
| 16 |
A |
1094 |
1087 |
34.50 |
|
|
|
| 17 |
A |
1036 |
1030 |
2.49 |
|
|
|
| 18 |
A |
977 |
971 |
12.06 |
|
|
|
| 19 |
A |
925 |
920 |
0.38 |
|
|
|
| 20 |
A |
831 |
826 |
7.06 |
|
|
|
| 21 |
A |
792 |
787 |
3.01 |
|
|
|
| 22 |
A |
763 |
758 |
0.63 |
|
|
|
| 23 |
A |
703 |
699 |
5.45 |
|
|
|
| 24 |
A |
649 |
645 |
0.88 |
|
|
|
| 25 |
A |
540 |
537 |
4.35 |
|
|
|
| 26 |
A |
468 |
465 |
3.84 |
|
|
|
| 27 |
A |
429 |
427 |
2.85 |
|
|
|
| 28 |
A |
413 |
411 |
3.83 |
|
|
|
| 29 |
A |
186 |
185 |
2.39 |
|
|
|
| 30 |
A |
62 |
62 |
9.45 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19874.9 cm
-1
Scaled (by 0.9942) Zero Point Vibrational Energy (zpe) 19759.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.029 |
|
|
|
| 2 |
C |
-0.012 |
|
|
|
| 3 |
H |
0.051 |
|
|
|
| 4 |
H |
0.074 |
|
|
|
| 5 |
C |
-0.130 |
|
|
|
| 6 |
H |
0.071 |
|
|
|
| 7 |
H |
0.065 |
|
|
|
| 8 |
C |
-0.104 |
|
|
|
| 9 |
S |
-0.054 |
|
|
|
| 10 |
H |
0.086 |
|
|
|
| 11 |
O |
-0.166 |
|
|
|
| 12 |
H |
0.089 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.900 |
1.167 |
0.263 |
1.497 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-52.244 |
1.052 |
0.342 |
| y |
1.052 |
-38.596 |
-0.026 |
| z |
0.342 |
-0.026 |
-41.951 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-11.970 |
1.052 |
0.342 |
| y |
1.052 |
8.501 |
-0.026 |
| z |
0.342 |
-0.026 |
3.469 |
|
| Polar |
|---|
| 3z2-r2 | 6.938 |
|---|
| x2-y2 | -13.648 |
|---|
| xy | 1.052 |
|---|
| xz | 0.342 |
|---|
| yz | -0.026 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.278 |
0.126 |
-0.233 |
| y |
0.126 |
9.737 |
0.054 |
| z |
-0.233 |
0.054 |
6.462 |
<r2> (average value of r
2) Å
2
| <r2> |
187.801 |
| (<r2>)1/2 |
13.704 |