Jump to
S1C2
Energy calculated at PBEPBE/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -578.381079 |
| Energy at 298.15K | -578.388605 |
| Nuclear repulsion energy | 157.080734 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3059 |
3041 |
20.50 |
|
|
|
| 2 |
A' |
2999 |
2981 |
29.85 |
|
|
|
| 3 |
A' |
2982 |
2965 |
7.92 |
|
|
|
| 4 |
A' |
2970 |
2953 |
21.56 |
|
|
|
| 5 |
A' |
1433 |
1425 |
5.10 |
|
|
|
| 6 |
A' |
1417 |
1408 |
0.74 |
|
|
|
| 7 |
A' |
1412 |
1404 |
1.01 |
|
|
|
| 8 |
A' |
1345 |
1337 |
0.85 |
|
|
|
| 9 |
A' |
1308 |
1301 |
4.39 |
|
|
|
| 10 |
A' |
1215 |
1208 |
25.40 |
|
|
|
| 11 |
A' |
1088 |
1082 |
0.34 |
|
|
|
| 12 |
A' |
1031 |
1025 |
1.07 |
|
|
|
| 13 |
A' |
889 |
884 |
15.59 |
|
|
|
| 14 |
A' |
721 |
717 |
36.05 |
|
|
|
| 15 |
A' |
353 |
351 |
3.08 |
|
|
|
| 16 |
A' |
225 |
224 |
1.87 |
|
|
|
| 17 |
A" |
3065 |
3047 |
30.30 |
|
|
|
| 18 |
A" |
3048 |
3030 |
16.42 |
|
|
|
| 19 |
A" |
3020 |
3003 |
0.60 |
|
|
|
| 20 |
A" |
1418 |
1410 |
7.54 |
|
|
|
| 21 |
A" |
1264 |
1257 |
0.00 |
|
|
|
| 22 |
A" |
1189 |
1182 |
0.13 |
|
|
|
| 23 |
A" |
1039 |
1032 |
1.29 |
|
|
|
| 24 |
A" |
837 |
832 |
0.21 |
|
|
|
| 25 |
A" |
736 |
732 |
4.38 |
|
|
|
| 26 |
A" |
226 |
224 |
0.03 |
|
|
|
| 27 |
A" |
117 |
116 |
1.20 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20201.7 cm
-1
Scaled (by 0.9942) Zero Point Vibrational Energy (zpe) 20084.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.586 |
0.000 |
| C2 |
0.915 |
-0.632 |
0.000 |
| C3 |
2.391 |
-0.221 |
0.000 |
| Cl4 |
-1.757 |
0.120 |
0.000 |
| H5 |
0.153 |
1.215 |
0.900 |
| H6 |
0.153 |
1.215 |
-0.900 |
| H7 |
0.687 |
-1.256 |
-0.890 |
| H8 |
0.687 |
-1.256 |
0.890 |
| H9 |
3.050 |
-1.112 |
0.000 |
| H10 |
2.647 |
0.383 |
-0.896 |
| H11 |
2.647 |
0.383 |
0.896 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5233 | 2.5240 | 1.8174 | 1.1081 | 1.1081 | 2.1576 | 2.1576 | 3.4910 | 2.8013 | 2.8013 |
C2 | 1.5233 | | 1.5326 | 2.7752 | 2.1909 | 2.1909 | 1.1104 | 1.1104 | 2.1884 | 2.1979 | 2.1979 | C3 | 2.5240 | 1.5326 | | 4.1620 | 2.8075 | 2.8075 | 2.1834 | 2.1834 | 1.1082 | 1.1100 | 1.1100 | Cl4 | 1.8174 | 2.7752 | 4.1620 | | 2.3778 | 2.3778 | 2.9419 | 2.9419 | 4.9620 | 4.5010 | 4.5010 | H5 | 1.1081 | 2.1909 | 2.8075 | 2.3778 | | 1.7991 | 3.0968 | 2.5273 | 3.8234 | 3.1832 | 2.6287 | H6 | 1.1081 | 2.1909 | 2.8075 | 2.3778 | 1.7991 | | 2.5273 | 3.0968 | 3.8234 | 2.6287 | 3.1832 | H7 | 2.1576 | 1.1104 | 2.1834 | 2.9419 | 3.0968 | 2.5273 | | 1.7802 | 2.5291 | 2.5544 | 3.1166 | H8 | 2.1576 | 1.1104 | 2.1834 | 2.9419 | 2.5273 | 3.0968 | 1.7802 | | 2.5291 | 3.1166 | 2.5544 | H9 | 3.4910 | 2.1884 | 1.1082 | 4.9620 | 3.8234 | 3.8234 | 2.5291 | 2.5291 | | 1.7893 | 1.7893 | H10 | 2.8013 | 2.1979 | 1.1100 | 4.5010 | 3.1832 | 2.6287 | 2.5544 | 3.1166 | 1.7893 | | 1.7910 | H11 | 2.8013 | 2.1979 | 1.1100 | 4.5010 | 2.6287 | 3.1832 | 3.1166 | 2.5544 | 1.7893 | 1.7910 | |
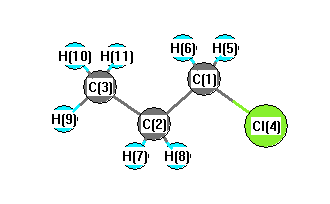 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
111.366 |
|
C1 |
C2 |
H7 |
109.008 |
| C1 |
C2 |
H8 |
109.008 |
|
C2 |
C1 |
Cl4 |
112.050 |
| C2 |
C1 |
H5 |
111.765 |
|
C2 |
C1 |
H6 |
111.765 |
| C2 |
C3 |
H9 |
110.909 |
|
C2 |
C3 |
H10 |
111.551 |
| C2 |
C3 |
H11 |
111.551 |
|
C3 |
C2 |
H7 |
110.382 |
| C3 |
C2 |
H8 |
110.382 |
|
Cl4 |
C1 |
H5 |
106.190 |
| Cl4 |
C1 |
H6 |
106.190 |
|
H5 |
C1 |
H6 |
108.552 |
| H7 |
C2 |
H8 |
106.562 |
|
H9 |
C3 |
H10 |
107.535 |
| H9 |
C3 |
H11 |
107.535 |
|
H10 |
C3 |
H11 |
107.558 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.095 |
|
|
|
| 2 |
C |
-0.046 |
|
|
|
| 3 |
C |
-0.070 |
|
|
|
| 4 |
Cl |
-0.136 |
|
|
|
| 5 |
H |
0.076 |
|
|
|
| 6 |
H |
0.076 |
|
|
|
| 7 |
H |
0.041 |
|
|
|
| 8 |
H |
0.041 |
|
|
|
| 9 |
H |
0.039 |
|
|
|
| 10 |
H |
0.038 |
|
|
|
| 11 |
H |
0.038 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.115 |
0.259 |
0.000 |
2.131 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.070 |
0.116 |
0.000 |
| y |
0.116 |
-32.466 |
0.000 |
| z |
0.000 |
0.000 |
-32.646 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.514 |
0.116 |
0.000 |
| y |
0.116 |
1.392 |
0.000 |
| z |
0.000 |
0.000 |
1.122 |
|
| Polar |
|---|
| 3z2-r2 | 2.243 |
|---|
| x2-y2 | -2.604 |
|---|
| xy | 0.116 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.893 |
0.015 |
0.000 |
| y |
0.015 |
5.925 |
0.000 |
| z |
0.000 |
0.000 |
5.507 |
<r2> (average value of r
2) Å
2
| <r2> |
154.385 |
| (<r2>)1/2 |
12.425 |
Jump to
S1C1
Energy calculated at PBEPBE/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -578.381411 |
| Energy at 298.15K | -578.389073 |
| Nuclear repulsion energy | 161.013840 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3071 |
3054 |
13.37 |
|
|
|
| 2 |
A |
3067 |
3049 |
15.25 |
|
|
|
| 3 |
A |
3053 |
3036 |
26.91 |
|
|
|
| 4 |
A |
3012 |
2995 |
23.34 |
|
|
|
| 5 |
A |
3000 |
2983 |
6.70 |
|
|
|
| 6 |
A |
2974 |
2956 |
20.25 |
|
|
|
| 7 |
A |
2956 |
2939 |
21.56 |
|
|
|
| 8 |
A |
1430 |
1422 |
5.00 |
|
|
|
| 9 |
A |
1421 |
1413 |
8.16 |
|
|
|
| 10 |
A |
1403 |
1395 |
1.49 |
|
|
|
| 11 |
A |
1396 |
1388 |
3.79 |
|
|
|
| 12 |
A |
1349 |
1341 |
6.08 |
|
|
|
| 13 |
A |
1320 |
1312 |
0.68 |
|
|
|
| 14 |
A |
1270 |
1263 |
24.51 |
|
|
|
| 15 |
A |
1225 |
1218 |
7.88 |
|
|
|
| 16 |
A |
1184 |
1177 |
0.51 |
|
|
|
| 17 |
A |
1080 |
1074 |
2.52 |
|
|
|
| 18 |
A |
1039 |
1033 |
0.84 |
|
|
|
| 19 |
A |
1016 |
1011 |
1.83 |
|
|
|
| 20 |
A |
882 |
877 |
11.01 |
|
|
|
| 21 |
A |
842 |
838 |
2.75 |
|
|
|
| 22 |
A |
767 |
763 |
19.04 |
|
|
|
| 23 |
A |
642 |
638 |
19.44 |
|
|
|
| 24 |
A |
413 |
410 |
1.52 |
|
|
|
| 25 |
A |
292 |
290 |
0.72 |
|
|
|
| 26 |
A |
211 |
210 |
1.42 |
|
|
|
| 27 |
A |
139 |
138 |
0.79 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20227.2 cm
-1
Scaled (by 0.9942) Zero Point Vibrational Energy (zpe) 20109.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/cc-pVDZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.177 |
0.889 |
0.307 |
| C2 |
-1.156 |
0.565 |
-0.354 |
| C3 |
-1.811 |
-0.725 |
0.143 |
| Cl4 |
1.462 |
-0.348 |
-0.068 |
| H5 |
0.584 |
1.857 |
-0.042 |
| H6 |
0.095 |
0.912 |
1.412 |
| H7 |
-1.825 |
1.434 |
-0.156 |
| H8 |
-1.013 |
0.527 |
-1.455 |
| H9 |
-2.780 |
-0.901 |
-0.364 |
| H10 |
-1.160 |
-1.600 |
-0.050 |
| H11 |
-2.005 |
-0.683 |
1.236 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5235 | 2.5665 | 1.8228 | 1.1065 | 1.1083 | 2.1256 | 2.1570 | 3.5219 | 2.8483 | 2.8456 |
C2 | 1.5235 | | 1.5293 | 2.7874 | 2.1906 | 2.1924 | 1.1139 | 1.1110 | 2.1876 | 2.1862 | 2.1918 | C3 | 2.5665 | 1.5293 | | 3.3014 | 3.5271 | 2.8149 | 2.1791 | 2.1817 | 1.1081 | 1.1083 | 1.1103 | Cl4 | 1.8228 | 2.7874 | 3.3014 | | 2.3740 | 2.3760 | 3.7394 | 2.9690 | 4.2882 | 2.9051 | 3.7189 | H5 | 1.1065 | 2.1906 | 3.5271 | 2.3740 | | 1.8018 | 2.4485 | 2.5135 | 4.3628 | 3.8726 | 3.8457 | H6 | 1.1083 | 2.1924 | 2.8149 | 2.3760 | 1.8018 | | 2.5333 | 3.0978 | 3.8356 | 3.1658 | 2.6431 | H7 | 2.1256 | 1.1139 | 2.1791 | 3.7394 | 2.4485 | 2.5333 | | 1.7798 | 2.5314 | 3.1077 | 2.5396 | H8 | 2.1570 | 1.1110 | 2.1817 | 2.9690 | 2.5135 | 3.0978 | 1.7798 | | 2.5207 | 2.5541 | 3.1128 | H9 | 3.5219 | 2.1876 | 1.1081 | 4.2882 | 4.3628 | 3.8356 | 2.5314 | 2.5207 | | 1.7929 | 1.7914 | H10 | 2.8483 | 2.1862 | 1.1083 | 2.9051 | 3.8726 | 3.1658 | 3.1077 | 2.5541 | 1.7929 | | 1.7912 | H11 | 2.8456 | 2.1918 | 1.1103 | 3.7189 | 3.8457 | 2.6431 | 2.5396 | 3.1128 | 1.7914 | 1.7912 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
114.424 |
|
C1 |
C2 |
H7 |
106.361 |
| C1 |
C2 |
H8 |
108.915 |
|
C2 |
C1 |
Cl4 |
112.509 |
| C2 |
C1 |
H5 |
111.823 |
|
C2 |
C1 |
H6 |
111.856 |
| C2 |
C3 |
H9 |
111.081 |
|
C2 |
C3 |
H10 |
110.954 |
| C2 |
C3 |
H11 |
111.280 |
|
C3 |
C2 |
H7 |
110.061 |
| C3 |
C2 |
H8 |
110.442 |
|
Cl4 |
C1 |
H5 |
105.664 |
| Cl4 |
C1 |
H6 |
105.721 |
|
H5 |
C1 |
H6 |
108.888 |
| H7 |
C2 |
H8 |
106.250 |
|
H9 |
C3 |
H10 |
107.981 |
| H9 |
C3 |
H11 |
107.710 |
|
H10 |
C3 |
H11 |
107.676 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.084 |
|
|
|
| 2 |
C |
-0.059 |
|
|
|
| 3 |
C |
-0.058 |
|
|
|
| 4 |
Cl |
-0.139 |
|
|
|
| 5 |
H |
0.074 |
|
|
|
| 6 |
H |
0.076 |
|
|
|
| 7 |
H |
0.034 |
|
|
|
| 8 |
H |
0.040 |
|
|
|
| 9 |
H |
0.034 |
|
|
|
| 10 |
H |
0.049 |
|
|
|
| 11 |
H |
0.033 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.593 |
1.206 |
0.271 |
2.016 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-34.460 |
0.204 |
0.125 |
| y |
0.204 |
-31.815 |
0.398 |
| z |
0.125 |
0.398 |
-32.693 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.206 |
0.204 |
0.125 |
| y |
0.204 |
1.761 |
0.398 |
| z |
0.125 |
0.398 |
0.445 |
|
| Polar |
|---|
| 3z2-r2 | 0.889 |
|---|
| x2-y2 | -2.645 |
|---|
| xy | 0.204 |
|---|
| xz | 0.125 |
|---|
| yz | 0.398 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.791 |
-0.504 |
-0.092 |
| y |
-0.504 |
6.663 |
0.135 |
| z |
-0.092 |
0.135 |
5.687 |
<r2> (average value of r
2) Å
2
| <r2> |
132.272 |
| (<r2>)1/2 |
11.501 |