Vibrational Frequencies calculated at PBEPBE/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3528 |
3508 |
20.94 |
|
|
|
| 2 |
A |
3065 |
3047 |
13.99 |
|
|
|
| 3 |
A |
3054 |
3037 |
17.03 |
|
|
|
| 4 |
A |
3006 |
2989 |
48.16 |
|
|
|
| 5 |
A |
2992 |
2975 |
11.00 |
|
|
|
| 6 |
A |
2983 |
2966 |
13.66 |
|
|
|
| 7 |
A |
2910 |
2894 |
72.21 |
|
|
|
| 8 |
A |
1780 |
1770 |
373.82 |
|
|
|
| 9 |
A |
1457 |
1448 |
4.19 |
|
|
|
| 10 |
A |
1419 |
1411 |
12.53 |
|
|
|
| 11 |
A |
1392 |
1384 |
15.90 |
|
|
|
| 12 |
A |
1383 |
1375 |
19.51 |
|
|
|
| 13 |
A |
1313 |
1306 |
10.70 |
|
|
|
| 14 |
A |
1284 |
1277 |
15.82 |
|
|
|
| 15 |
A |
1246 |
1238 |
39.36 |
|
|
|
| 16 |
A |
1212 |
1205 |
42.27 |
|
|
|
| 17 |
A |
1194 |
1187 |
7.04 |
|
|
|
| 18 |
A |
1161 |
1154 |
2.22 |
|
|
|
| 19 |
A |
1133 |
1126 |
3.43 |
|
|
|
| 20 |
A |
1058 |
1052 |
12.14 |
|
|
|
| 21 |
A |
1040 |
1034 |
2.13 |
|
|
|
| 22 |
A |
983 |
977 |
10.78 |
|
|
|
| 23 |
A |
910 |
904 |
0.26 |
|
|
|
| 24 |
A |
878 |
873 |
1.72 |
|
|
|
| 25 |
A |
867 |
862 |
2.88 |
|
|
|
| 26 |
A |
789 |
785 |
5.62 |
|
|
|
| 27 |
A |
673 |
669 |
7.09 |
|
|
|
| 28 |
A |
614 |
610 |
14.98 |
|
|
|
| 29 |
A |
545 |
542 |
32.62 |
|
|
|
| 30 |
A |
478 |
475 |
54.60 |
|
|
|
| 31 |
A |
452 |
449 |
8.73 |
|
|
|
| 32 |
A |
200 |
198 |
3.50 |
|
|
|
| 33 |
A |
140 |
139 |
1.18 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23569.0 cm
-1
Scaled (by 0.9942) Zero Point Vibrational Energy (zpe) 23432.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
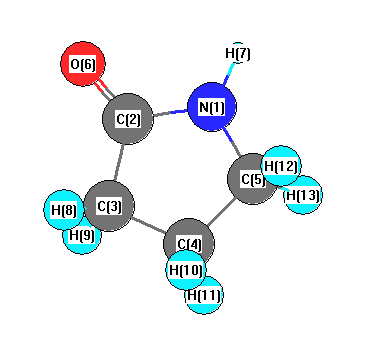
Charges from optimized geometry at PBEPBE/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.127 |
|
|
|
| 2 |
C |
0.085 |
|
|
|
| 3 |
C |
-0.049 |
|
|
|
| 4 |
C |
-0.053 |
|
|
|
| 5 |
C |
0.063 |
|
|
|
| 6 |
O |
-0.235 |
|
|
|
| 7 |
H |
0.085 |
|
|
|
| 8 |
H |
0.054 |
|
|
|
| 9 |
H |
0.044 |
|
|
|
| 10 |
H |
0.025 |
|
|
|
| 11 |
H |
0.041 |
|
|
|
| 12 |
H |
0.033 |
|
|
|
| 13 |
H |
0.033 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-3.535 |
-0.747 |
0.325 |
3.628 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.831 |
-0.234 |
0.205 |
| y |
-0.234 |
-31.542 |
-0.163 |
| z |
0.205 |
-0.163 |
-35.449 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-9.335 |
-0.234 |
0.205 |
| y |
-0.234 |
7.598 |
-0.163 |
| z |
0.205 |
-0.163 |
1.737 |
|
| Polar |
|---|
| 3z2-r2 | 3.474 |
|---|
| x2-y2 | -11.289 |
|---|
| xy | -0.234 |
|---|
| xz | 0.205 |
|---|
| yz | -0.163 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.075 |
0.429 |
-0.098 |
| y |
0.429 |
8.091 |
0.029 |
| z |
-0.098 |
0.029 |
5.873 |
<r2> (average value of r
2) Å
2
| <r2> |
147.357 |
| (<r2>)1/2 |
12.139 |