Vibrational Frequencies calculated at PBEPBE/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3052 |
3020 |
35.79 |
|
|
|
| 2 |
A' |
3042 |
3011 |
37.56 |
|
|
|
| 3 |
A' |
2973 |
2942 |
38.91 |
|
|
|
| 4 |
A' |
2965 |
2934 |
23.95 |
|
|
|
| 5 |
A' |
2891 |
2861 |
95.48 |
|
|
|
| 6 |
A' |
2860 |
2830 |
48.73 |
|
|
|
| 7 |
A' |
1495 |
1479 |
3.66 |
|
|
|
| 8 |
A' |
1477 |
1462 |
10.85 |
|
|
|
| 9 |
A' |
1470 |
1455 |
6.02 |
|
|
|
| 10 |
A' |
1457 |
1442 |
2.94 |
|
|
|
| 11 |
A' |
1442 |
1427 |
0.12 |
|
|
|
| 12 |
A' |
1388 |
1373 |
20.22 |
|
|
|
| 13 |
A' |
1377 |
1363 |
1.95 |
|
|
|
| 14 |
A' |
1296 |
1282 |
1.49 |
|
|
|
| 15 |
A' |
1197 |
1185 |
23.13 |
|
|
|
| 16 |
A' |
1129 |
1118 |
134.04 |
|
|
|
| 17 |
A' |
1102 |
1091 |
29.76 |
|
|
|
| 18 |
A' |
1041 |
1030 |
2.50 |
|
|
|
| 19 |
A' |
960 |
950 |
24.57 |
|
|
|
| 20 |
A' |
890 |
881 |
7.89 |
|
|
|
| 21 |
A' |
430 |
425 |
0.49 |
|
|
|
| 22 |
A' |
400 |
396 |
2.94 |
|
|
|
| 23 |
A' |
189 |
187 |
1.30 |
|
|
|
| 24 |
A" |
3034 |
3002 |
70.20 |
|
|
|
| 25 |
A" |
3005 |
2973 |
5.08 |
|
|
|
| 26 |
A" |
2930 |
2899 |
78.92 |
|
|
|
| 27 |
A" |
2883 |
2853 |
82.66 |
|
|
|
| 28 |
A" |
1473 |
1458 |
9.87 |
|
|
|
| 29 |
A" |
1449 |
1434 |
8.38 |
|
|
|
| 30 |
A" |
1286 |
1272 |
0.28 |
|
|
|
| 31 |
A" |
1238 |
1225 |
1.86 |
|
|
|
| 32 |
A" |
1167 |
1155 |
4.82 |
|
|
|
| 33 |
A" |
1140 |
1128 |
0.06 |
|
|
|
| 34 |
A" |
881 |
872 |
2.32 |
|
|
|
| 35 |
A" |
749 |
741 |
2.10 |
|
|
|
| 36 |
A" |
243 |
241 |
2.34 |
|
|
|
| 37 |
A" |
238 |
236 |
0.92 |
|
|
|
| 38 |
A" |
112 |
111 |
2.93 |
|
|
|
| 39 |
A" |
104 |
103 |
0.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29225.7 cm
-1
Scaled (by 0.9896) Zero Point Vibrational Energy (zpe) 28921.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
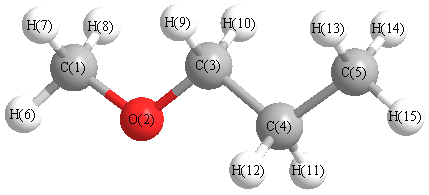
Charges from optimized geometry at PBEPBE/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.458 |
|
|
|
| 2 |
O |
-0.289 |
|
|
|
| 3 |
C |
-0.203 |
|
|
|
| 4 |
C |
-0.448 |
|
|
|
| 5 |
C |
-0.648 |
|
|
|
| 6 |
H |
0.218 |
|
|
|
| 7 |
H |
0.190 |
|
|
|
| 8 |
H |
0.190 |
|
|
|
| 9 |
H |
0.181 |
|
|
|
| 10 |
H |
0.181 |
|
|
|
| 11 |
H |
0.218 |
|
|
|
| 12 |
H |
0.218 |
|
|
|
| 13 |
H |
0.213 |
|
|
|
| 14 |
H |
0.213 |
|
|
|
| 15 |
H |
0.222 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.198 |
0.991 |
0.000 |
1.010 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.630 |
-2.078 |
0.000 |
| y |
-2.078 |
-34.038 |
0.000 |
| z |
0.000 |
0.000 |
-33.554 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.166 |
-2.078 |
0.000 |
| y |
-2.078 |
-1.446 |
0.000 |
| z |
0.000 |
0.000 |
-0.720 |
|
| Polar |
|---|
| 3z2-r2 | -1.440 |
|---|
| x2-y2 | 2.408 |
|---|
| xy | -2.078 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.512 |
-0.479 |
0.000 |
| y |
-0.479 |
7.367 |
0.000 |
| z |
0.000 |
0.000 |
7.030 |
<r2> (average value of r
2) Å
2
| <r2> |
182.466 |
| (<r2>)1/2 |
13.508 |