Jump to
S1C2
Vibrational Frequencies calculated at PBEPBE/6-311G*
Geometric Data calculated at PBEPBE/6-311G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at PBEPBE/6-311G*
| | hartrees |
|---|
| Energy at 0K | -232.220157 |
| Energy at 298.15K | -232.228261 |
| Nuclear repulsion energy | 175.168387 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3085 |
3053 |
13.31 |
|
|
|
| 2 |
A |
3063 |
3031 |
24.09 |
|
|
|
| 3 |
A |
3056 |
3024 |
26.90 |
|
|
|
| 4 |
A |
3032 |
3000 |
13.21 |
|
|
|
| 5 |
A |
2987 |
2956 |
24.97 |
|
|
|
| 6 |
A |
2974 |
2943 |
14.17 |
|
|
|
| 7 |
A |
2970 |
2939 |
5.93 |
|
|
|
| 8 |
A |
2947 |
2916 |
19.47 |
|
|
|
| 9 |
A |
1742 |
1723 |
131.75 |
|
|
|
| 10 |
A |
1470 |
1455 |
10.34 |
|
|
|
| 11 |
A |
1462 |
1447 |
10.59 |
|
|
|
| 12 |
A |
1444 |
1429 |
12.12 |
|
|
|
| 13 |
A |
1434 |
1419 |
21.76 |
|
|
|
| 14 |
A |
1416 |
1401 |
8.75 |
|
|
|
| 15 |
A |
1378 |
1363 |
9.79 |
|
|
|
| 16 |
A |
1345 |
1331 |
59.24 |
|
|
|
| 17 |
A |
1327 |
1313 |
15.18 |
|
|
|
| 18 |
A |
1251 |
1238 |
0.00 |
|
|
|
| 19 |
A |
1153 |
1141 |
71.72 |
|
|
|
| 20 |
A |
1095 |
1083 |
0.49 |
|
|
|
| 21 |
A |
1081 |
1069 |
0.83 |
|
|
|
| 22 |
A |
986 |
975 |
5.10 |
|
|
|
| 23 |
A |
926 |
916 |
4.30 |
|
|
|
| 24 |
A |
913 |
903 |
12.79 |
|
|
|
| 25 |
A |
745 |
737 |
4.66 |
|
|
|
| 26 |
A |
729 |
721 |
3.04 |
|
|
|
| 27 |
A |
575 |
569 |
5.70 |
|
|
|
| 28 |
A |
460 |
456 |
0.32 |
|
|
|
| 29 |
A |
390 |
386 |
3.88 |
|
|
|
| 30 |
A |
242 |
239 |
4.94 |
|
|
|
| 31 |
A |
187 |
185 |
0.18 |
|
|
|
| 32 |
A |
105 |
104 |
0.08 |
|
|
|
| 33 |
A |
53 |
53 |
0.12 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24009.3 cm
-1
Scaled (by 0.9896) Zero Point Vibrational Energy (zpe) 23759.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
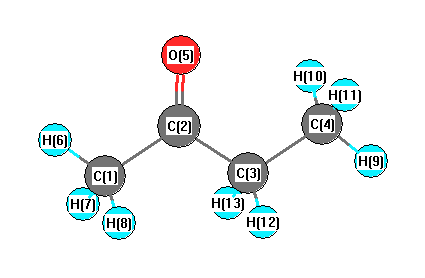
Geometric Data calculated at PBEPBE/6-311G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.890 |
-0.507 |
0.033 |
| C2 |
0.529 |
0.170 |
-0.020 |
| C3 |
-0.686 |
-0.755 |
-0.059 |
| C4 |
-2.020 |
-0.024 |
0.048 |
| O5 |
0.414 |
1.385 |
-0.023 |
| H6 |
2.681 |
0.228 |
-0.159 |
| H7 |
2.048 |
-0.940 |
1.036 |
| H8 |
1.963 |
-1.338 |
-0.686 |
| H9 |
-2.863 |
-0.727 |
-0.020 |
| H10 |
-2.100 |
0.517 |
1.002 |
| H11 |
-2.124 |
0.722 |
-0.752 |
| H12 |
-0.626 |
-1.336 |
-0.999 |
| H13 |
-0.572 |
-1.513 |
0.740 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5209 | 2.5887 | 3.9388 | 2.3994 | 1.0972 | 1.1036 | 1.1018 | 4.7581 | 4.2308 | 4.2704 | 2.8429 | 2.7516 |
C2 | 1.5209 | | 1.5269 | 2.5564 | 1.2202 | 2.1582 | 2.1580 | 2.1856 | 3.5083 | 2.8411 | 2.8068 | 2.1357 | 2.1495 | C3 | 2.5887 | 1.5269 | | 1.5250 | 2.4064 | 3.5091 | 2.9507 | 2.7840 | 2.1781 | 2.1780 | 2.1755 | 1.1072 | 1.1063 | C4 | 3.9388 | 2.5564 | 1.5250 | | 2.8131 | 4.7123 | 4.2853 | 4.2580 | 1.1003 | 1.0999 | 1.0992 | 2.1820 | 2.1886 | O5 | 2.3994 | 1.2202 | 2.4064 | 2.8131 | | 2.5488 | 3.0326 | 3.2022 | 3.8989 | 2.8500 | 2.7231 | 3.0725 | 3.1545 | H6 | 1.0972 | 2.1582 | 3.5091 | 4.7123 | 2.5488 | | 1.7869 | 1.8016 | 5.6280 | 4.9285 | 4.8671 | 3.7539 | 3.7979 | H7 | 1.1036 | 2.1580 | 2.9507 | 4.2853 | 3.0326 | 1.7869 | | 1.7688 | 5.0283 | 4.3968 | 4.8343 | 3.3838 | 2.6988 | H8 | 1.1018 | 2.1856 | 2.7840 | 4.2580 | 3.2022 | 1.8016 | 1.7688 | | 4.9105 | 4.7749 | 4.5778 | 2.6083 | 2.9140 | H9 | 4.7581 | 3.5083 | 2.1781 | 1.1003 | 3.8989 | 5.6280 | 5.0283 | 4.9105 | | 1.7822 | 1.7840 | 2.5169 | 2.5383 | H10 | 4.2308 | 2.8411 | 2.1780 | 1.0999 | 2.8500 | 4.9285 | 4.3968 | 4.7749 | 1.7822 | | 1.7667 | 3.1004 | 2.5540 | H11 | 4.2704 | 2.8068 | 2.1755 | 1.0992 | 2.7231 | 4.8671 | 4.8343 | 4.5778 | 1.7840 | 1.7667 | | 2.5578 | 3.1031 | H12 | 2.8429 | 2.1357 | 1.1072 | 2.1820 | 3.0725 | 3.7539 | 3.3838 | 2.6083 | 2.5169 | 3.1004 | 2.5578 | | 1.7484 | H13 | 2.7516 | 2.1495 | 1.1063 | 2.1886 | 3.1545 | 3.7979 | 2.6988 | 2.9140 | 2.5383 | 2.5540 | 3.1031 | 1.7484 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
116.287 |
|
C1 |
C2 |
O5 |
121.792 |
| C2 |
C1 |
H6 |
109.987 |
|
C2 |
C1 |
H7 |
109.597 |
| C2 |
C1 |
H8 |
111.902 |
|
C2 |
C3 |
C4 |
113.781 |
| C2 |
C3 |
H12 |
107.267 |
|
C2 |
C3 |
H13 |
108.368 |
| C3 |
C2 |
O5 |
121.919 |
|
C3 |
C4 |
H9 |
111.097 |
| C3 |
C4 |
H10 |
111.111 |
|
C3 |
C4 |
H11 |
110.957 |
| C4 |
C3 |
H12 |
110.988 |
|
C4 |
C3 |
H13 |
111.569 |
| H6 |
C1 |
H7 |
108.567 |
|
H6 |
C1 |
H8 |
110.026 |
| H7 |
C1 |
H8 |
106.657 |
|
H9 |
C4 |
H10 |
108.199 |
| H9 |
C4 |
H11 |
108.407 |
|
H10 |
C4 |
H11 |
106.914 |
| H12 |
C3 |
H13 |
104.347 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.714 |
|
|
|
| 2 |
C |
0.296 |
|
|
|
| 3 |
C |
-0.516 |
|
|
|
| 4 |
C |
-0.637 |
|
|
|
| 5 |
O |
-0.266 |
|
|
|
| 6 |
H |
0.241 |
|
|
|
| 7 |
H |
0.239 |
|
|
|
| 8 |
H |
0.228 |
|
|
|
| 9 |
H |
0.213 |
|
|
|
| 10 |
H |
0.225 |
|
|
|
| 11 |
H |
0.229 |
|
|
|
| 12 |
H |
0.234 |
|
|
|
| 13 |
H |
0.228 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.221 |
-2.627 |
0.037 |
2.636 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.601 |
-1.008 |
0.178 |
| y |
-1.008 |
-35.140 |
0.030 |
| z |
0.178 |
0.030 |
-30.860 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.399 |
-1.008 |
0.178 |
| y |
-1.008 |
-4.410 |
0.030 |
| z |
0.178 |
0.030 |
2.011 |
|
| Polar |
|---|
| 3z2-r2 | 4.021 |
|---|
| x2-y2 | 4.539 |
|---|
| xy | -1.008 |
|---|
| xz | 0.178 |
|---|
| yz | 0.030 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.482 |
-0.204 |
0.046 |
| y |
-0.204 |
7.624 |
0.002 |
| z |
0.046 |
0.002 |
5.859 |
<r2> (average value of r
2) Å
2
| <r2> |
138.285 |
| (<r2>)1/2 |
11.759 |