All results from a given calculation for C4H12N2 (1,2-Butanediamine)
using model chemistry: MP3/6-31+G**
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
C1 |
1A |
Energy calculated at MP3/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -268.356839 |
| Energy at 298.15K | |
| HF Energy | -267.363988 |
| Nuclear repulsion energy | 263.539721 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP3/6-31+G**
Geometric Data calculated at MP3/6-31+G**
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
1.934 |
-0.637 |
-0.640 |
| H2 |
1.847 |
0.136 |
-1.286 |
| H3 |
2.914 |
-0.876 |
-0.573 |
| C4 |
-2.400 |
-0.344 |
-0.099 |
| H5 |
-3.020 |
-1.165 |
-0.456 |
| H6 |
-2.747 |
-0.071 |
0.899 |
| H7 |
-2.579 |
0.504 |
-0.760 |
| N8 |
0.025 |
1.491 |
-0.344 |
| H9 |
0.506 |
2.245 |
0.132 |
| H10 |
-0.914 |
1.819 |
-0.523 |
| C11 |
-0.927 |
-0.753 |
-0.078 |
| H12 |
-0.816 |
-1.666 |
0.510 |
| H13 |
-0.583 |
-0.983 |
-1.085 |
| C14 |
1.428 |
-0.206 |
0.658 |
| H15 |
2.024 |
0.589 |
1.133 |
| H16 |
1.434 |
-1.056 |
1.343 |
| C17 |
-0.004 |
0.306 |
0.521 |
| H18 |
-0.365 |
0.530 |
1.536 |
Atom - Atom Distances (Å)
| |
N1 |
H2 |
H3 |
C4 |
H5 |
H6 |
H7 |
N8 |
H9 |
H10 |
C11 |
H12 |
H13 |
C14 |
H15 |
H16 |
C17 |
H18 |
| N1 | | 1.0117 | 1.0104 | 4.3776 | 4.9857 | 4.9599 | 4.6565 | 2.8750 | 3.3076 | 3.7635 | 2.9183 | 3.1534 | 2.5801 | 1.4580 | 2.1576 | 2.0871 | 2.4484 | 3.3746 |
H2 | 1.0117 | | 1.6348 | 4.4353 | 5.1053 | 5.0908 | 4.4714 | 2.4588 | 2.8733 | 3.3225 | 3.1533 | 3.6827 | 2.6826 | 2.0179 | 2.4683 | 2.9161 | 2.5925 | 3.6079 | H3 | 1.0104 | 1.6348 | | 5.3612 | 5.9420 | 5.9039 | 5.6662 | 3.7423 | 4.0041 | 4.6822 | 3.8746 | 3.9631 | 3.5362 | 2.0422 | 2.4185 | 2.4271 | 3.3329 | 4.1449 | C4 | 4.3776 | 4.4353 | 5.3612 | | 1.0890 | 1.0907 | 1.0903 | 3.0507 | 3.8985 | 2.6582 | 1.5287 | 2.1514 | 2.1634 | 3.9045 | 4.6857 | 4.1575 | 2.5583 | 2.7526 | H5 | 4.9857 | 5.1053 | 5.9420 | 1.0890 | | 1.7619 | 1.7528 | 4.0421 | 4.9400 | 3.6527 | 2.1661 | 2.4582 | 2.5231 | 4.6846 | 5.5713 | 4.8048 | 3.4945 | 3.7268 | H6 | 4.9599 | 5.0908 | 5.9039 | 1.0907 | 1.7619 | | 1.7642 | 3.4158 | 4.0663 | 2.9926 | 2.1750 | 2.5345 | 3.0735 | 4.1841 | 4.8217 | 4.3183 | 2.7941 | 2.5378 | H7 | 4.6565 | 4.4714 | 5.6662 | 1.0903 | 1.7528 | 1.7642 | | 2.8154 | 3.6527 | 2.1344 | 2.1849 | 3.0711 | 2.5094 | 4.3093 | 4.9775 | 4.7917 | 2.8826 | 3.1898 | N8 | 2.8750 | 2.4588 | 3.7423 | 3.0507 | 4.0421 | 3.4158 | 2.8154 | | 1.0125 | 1.0109 | 2.4527 | 3.3774 | 2.6537 | 2.4195 | 2.6444 | 3.3647 | 1.4679 | 2.1472 | H9 | 3.3076 | 2.8733 | 4.0041 | 3.8985 | 4.9400 | 4.0663 | 3.6527 | 1.0125 | | 1.6213 | 3.3296 | 4.1456 | 3.6174 | 2.6707 | 2.4594 | 3.6365 | 2.0425 | 2.3814 | H10 | 3.7635 | 3.3225 | 4.6822 | 2.6582 | 3.6527 | 2.9926 | 2.1344 | 1.0109 | 1.6213 | | 2.6109 | 3.6369 | 2.8771 | 3.3144 | 3.5907 | 4.1555 | 2.0521 | 2.4912 | C11 | 2.9183 | 3.1533 | 3.8746 | 1.5287 | 2.1661 | 2.1750 | 2.1849 | 2.4527 | 3.3296 | 2.6109 | | 1.0917 | 1.0885 | 2.5276 | 3.4607 | 2.7724 | 1.5272 | 2.1378 | H12 | 3.1534 | 3.6827 | 3.9631 | 2.1514 | 2.4582 | 2.5345 | 3.0711 | 3.3774 | 4.1456 | 3.6369 | 1.0917 | | 1.7508 | 2.6814 | 3.6792 | 2.4754 | 2.1325 | 2.4662 | H13 | 2.5801 | 2.6826 | 3.5362 | 2.1634 | 2.5231 | 3.0735 | 2.5094 | 2.6537 | 3.6174 | 2.8771 | 1.0885 | 1.7508 | | 2.7727 | 3.7669 | 3.1576 | 2.1391 | 3.0348 | C14 | 1.4580 | 2.0179 | 2.0422 | 3.9045 | 4.6846 | 4.1841 | 4.3093 | 2.4195 | 2.6707 | 3.3144 | 2.5276 | 2.6814 | 2.7727 | | 1.1011 | 1.0919 | 1.5271 | 2.1287 | H15 | 2.1576 | 2.4683 | 2.4185 | 4.6857 | 5.5713 | 4.8217 | 4.9775 | 2.6444 | 2.4594 | 3.5907 | 3.4607 | 3.6792 | 3.7669 | 1.1011 | | 1.7599 | 2.1372 | 2.4236 | H16 | 2.0871 | 2.9161 | 2.4271 | 4.1575 | 4.8048 | 4.3183 | 4.7917 | 3.3647 | 3.6365 | 4.1555 | 2.7724 | 2.4754 | 3.1576 | 1.0919 | 1.7599 | | 2.1445 | 2.4070 | C17 | 2.4484 | 2.5925 | 3.3329 | 2.5583 | 3.4945 | 2.7941 | 2.8826 | 1.4679 | 2.0425 | 2.0521 | 1.5272 | 2.1325 | 2.1391 | 1.5271 | 2.1372 | 2.1445 | | 1.1009 | H18 | 3.3746 | 3.6079 | 4.1449 | 2.7526 | 3.7268 | 2.5378 | 3.1898 | 2.1472 | 2.3814 | 2.4912 | 2.1378 | 2.4662 | 3.0348 | 2.1287 | 2.4236 | 2.4070 | 1.1009 | |
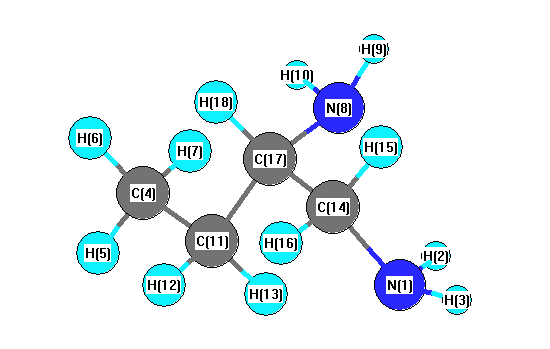 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C14 |
H15 |
114.216 |
|
N1 |
C14 |
H16 |
109.025 |
| N1 |
C14 |
C17 |
110.182 |
|
H2 |
N1 |
H3 |
107.889 |
| H2 |
N1 |
C14 |
108.227 |
|
H3 |
N1 |
C14 |
110.330 |
| C4 |
C11 |
H12 |
109.242 |
|
C4 |
C11 |
H13 |
110.373 |
| C4 |
C11 |
C17 |
113.690 |
|
H5 |
C4 |
H6 |
107.864 |
| H5 |
C4 |
H7 |
107.088 |
|
H5 |
C4 |
C11 |
110.564 |
| H6 |
C4 |
H7 |
107.977 |
|
H6 |
C4 |
C11 |
111.165 |
| H7 |
C4 |
C11 |
111.994 |
|
N8 |
C17 |
C11 |
109.934 |
| N8 |
C17 |
C14 |
107.752 |
|
N8 |
C17 |
H18 |
112.633 |
| H9 |
N8 |
H10 |
106.504 |
|
H9 |
N8 |
C17 |
109.491 |
| H10 |
N8 |
C17 |
110.397 |
|
C11 |
C17 |
C14 |
111.693 |
| C11 |
C17 |
H18 |
107.767 |
|
H12 |
C11 |
H13 |
106.841 |
| H12 |
C11 |
C17 |
107.874 |
|
H13 |
C11 |
C17 |
108.568 |
| C14 |
C17 |
H18 |
107.075 |
|
H15 |
C14 |
H16 |
106.745 |
| H15 |
C14 |
C17 |
107.709 |
|
H16 |
C14 |
C17 |
108.797 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability