Jump to
S1C2
Energy calculated at CCSD=FULL/6-31G*
| | hartrees |
|---|
| Energy at 0K | -490.831815 |
| Energy at 298.15K | -490.832526 |
| HF Energy | -490.389640 |
| Nuclear repulsion energy | 79.507422 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD=FULL/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3723 |
3536 |
251.95 |
|
|
|
| 2 |
A' |
2091 |
1986 |
750.34 |
|
|
|
| 3 |
A' |
888 |
844 |
6.06 |
|
|
|
| 4 |
A' |
621 |
590 |
445.23 |
|
|
|
| 5 |
A' |
445 |
423 |
126.46 |
|
|
|
| 6 |
A" |
476 |
453 |
1.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4122.4 cm
-1
Scaled (by 0.9498) Zero Point Vibrational Energy (zpe) 3915.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
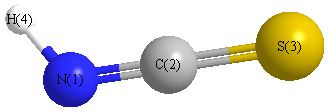
Geometric Data calculated at CCSD=FULL/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-0.162 |
1.693 |
0.000 |
| C2 |
0.000 |
0.497 |
0.000 |
| S3 |
0.041 |
-1.081 |
0.000 |
| H4 |
0.485 |
2.467 |
0.000 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
S3 |
H4 |
| N1 | | 1.2065 | 2.7814 | 1.0088 |
C2 | 1.2065 | | 1.5790 | 2.0283 | S3 | 2.7814 | 1.5790 | | 3.5757 | H4 | 1.0088 | 2.0283 | 3.5757 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
S3 |
173.746 |
|
C2 |
N1 |
H4 |
132.377 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCSD=FULL/6-31G*
| | hartrees |
|---|
| Energy at 0K | -490.828444 |
| Energy at 298.15K | |
| HF Energy | -490.389119 |
| Nuclear repulsion energy | 79.514520 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD=FULL/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3927 |
3730 |
743.20 |
|
|
|
| 2 |
Σ |
2222 |
2111 |
714.59 |
|
|
|
| 3 |
Σ |
858 |
815 |
49.09 |
|
|
|
| 4 |
Π |
473 |
449 |
5.59 |
|
|
|
| 4 |
Π |
473 |
449 |
5.59 |
|
|
|
| 5 |
Π |
432i |
411i |
188.77 |
|
|
|
| 5 |
Π |
432i |
411i |
188.77 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3543.5 cm
-1
Scaled (by 0.9498) Zero Point Vibrational Energy (zpe) 3365.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD=FULL/6-31G*
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
-1.682 |
| C2 |
0.000 |
0.000 |
-0.502 |
| S3 |
0.000 |
0.000 |
1.091 |
| H4 |
0.000 |
0.000 |
-2.678 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
S3 |
H4 |
| N1 | | 1.1801 | 2.7733 | 0.9961 |
C2 | 1.1801 | | 1.5932 | 2.1762 | S3 | 2.7733 | 1.5932 | | 3.7694 | H4 | 0.9961 | 2.1762 | 3.7694 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
S3 |
180.000 |
|
C2 |
N1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability