Jump to
S1C2
Vibrational Frequencies calculated at B3LYPultrafine/6-31G*
Geometric Data calculated at B3LYPultrafine/6-31G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B3LYPultrafine/6-31G*
| | hartrees |
|---|
| Energy at 0K | -232.470556 |
| Energy at 298.15K | -232.478741 |
| Nuclear repulsion energy | 175.510382 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYPultrafine/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3165 |
3032 |
11.08 |
|
|
|
| 2 |
A |
3141 |
3009 |
24.37 |
|
|
|
| 3 |
A |
3133 |
3002 |
24.28 |
|
|
|
| 4 |
A |
3106 |
2975 |
12.86 |
|
|
|
| 5 |
A |
3066 |
2937 |
22.52 |
|
|
|
| 6 |
A |
3051 |
2922 |
11.63 |
|
|
|
| 7 |
A |
3049 |
2921 |
8.14 |
|
|
|
| 8 |
A |
3024 |
2897 |
16.83 |
|
|
|
| 9 |
A |
1817 |
1740 |
128.94 |
|
|
|
| 10 |
A |
1529 |
1465 |
7.80 |
|
|
|
| 11 |
A |
1525 |
1461 |
5.79 |
|
|
|
| 12 |
A |
1506 |
1443 |
9.87 |
|
|
|
| 13 |
A |
1497 |
1434 |
17.57 |
|
|
|
| 14 |
A |
1484 |
1422 |
2.51 |
|
|
|
| 15 |
A |
1443 |
1382 |
4.62 |
|
|
|
| 16 |
A |
1413 |
1354 |
37.03 |
|
|
|
| 17 |
A |
1390 |
1332 |
13.15 |
|
|
|
| 18 |
A |
1297 |
1242 |
0.05 |
|
|
|
| 19 |
A |
1200 |
1149 |
77.39 |
|
|
|
| 20 |
A |
1145 |
1097 |
0.40 |
|
|
|
| 21 |
A |
1118 |
1071 |
1.92 |
|
|
|
| 22 |
A |
1007 |
965 |
2.44 |
|
|
|
| 23 |
A |
963 |
923 |
4.08 |
|
|
|
| 24 |
A |
949 |
910 |
10.93 |
|
|
|
| 25 |
A |
765 |
733 |
2.49 |
|
|
|
| 26 |
A |
765 |
733 |
3.21 |
|
|
|
| 27 |
A |
589 |
564 |
8.72 |
|
|
|
| 28 |
A |
474 |
454 |
0.03 |
|
|
|
| 29 |
A |
401 |
384 |
4.07 |
|
|
|
| 30 |
A |
249 |
239 |
5.04 |
|
|
|
| 31 |
A |
205 |
196 |
0.18 |
|
|
|
| 32 |
A |
106 |
102 |
0.00 |
|
|
|
| 33 |
A |
30 |
29 |
0.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24800.7 cm
-1
Scaled (by 0.958) Zero Point Vibrational Energy (zpe) 23759.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYPultrafine/6-31G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-1.894 |
-0.499 |
0.000 |
| C2 |
-0.527 |
0.168 |
-0.000 |
| C3 |
0.684 |
-0.760 |
-0.000 |
| C4 |
2.020 |
-0.020 |
0.000 |
| O5 |
-0.409 |
1.378 |
-0.000 |
| H6 |
-2.677 |
0.262 |
0.000 |
| H7 |
-2.009 |
-1.143 |
-0.881 |
| H8 |
-2.009 |
-1.143 |
0.881 |
| H9 |
2.855 |
-0.729 |
-0.000 |
| H10 |
2.111 |
0.623 |
-0.880 |
| H11 |
2.111 |
0.622 |
0.882 |
| H12 |
0.599 |
-1.426 |
0.872 |
| H13 |
0.600 |
-1.425 |
-0.873 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5209 | 2.5914 | 3.9436 | 2.3940 | 1.0918 | 1.0973 | 1.0973 | 4.7548 | 4.2514 | 4.2513 | 2.7993 | 2.7997 |
C2 | 1.5209 | | 1.5256 | 2.5545 | 1.2165 | 2.1518 | 2.1655 | 2.1657 | 3.4992 | 2.8181 | 2.8185 | 2.1373 | 2.1372 | C3 | 2.5914 | 1.5256 | | 1.5271 | 2.4013 | 3.5130 | 2.8594 | 2.8596 | 2.1712 | 2.1729 | 2.1729 | 1.1004 | 1.1004 | C4 | 3.9436 | 2.5545 | 1.5271 | | 2.8031 | 4.7058 | 4.2747 | 4.2746 | 1.0950 | 1.0940 | 1.0940 | 2.1800 | 2.1800 | O5 | 2.3940 | 1.2165 | 2.4013 | 2.8031 | | 2.5281 | 3.1135 | 3.1136 | 3.8852 | 2.7739 | 2.7749 | 3.1047 | 3.1042 | H6 | 1.0918 | 2.1518 | 3.5130 | 4.7058 | 2.5281 | | 1.7877 | 1.7878 | 5.6202 | 4.8816 | 4.8818 | 3.7872 | 3.7873 | H7 | 1.0973 | 2.1655 | 2.8594 | 4.2747 | 3.1135 | 1.7877 | | 1.7623 | 4.9606 | 4.4826 | 4.8161 | 3.1554 | 2.6241 | H8 | 1.0973 | 2.1657 | 2.8596 | 4.2746 | 3.1136 | 1.7878 | 1.7623 | | 4.9607 | 4.8161 | 4.4819 | 2.6239 | 3.1564 | H9 | 4.7548 | 3.4992 | 2.1712 | 1.0950 | 3.8852 | 5.6202 | 4.9606 | 4.9607 | | 1.7763 | 1.7762 | 2.5166 | 2.5164 | H10 | 4.2514 | 2.8181 | 2.1729 | 1.0940 | 2.7739 | 4.8816 | 4.4826 | 4.8161 | 1.7763 | | 1.7615 | 3.0901 | 2.5449 | H11 | 4.2513 | 2.8185 | 2.1729 | 1.0940 | 2.7749 | 4.8818 | 4.8161 | 4.4819 | 1.7762 | 1.7615 | | 2.5447 | 3.0902 | H12 | 2.7993 | 2.1373 | 1.1004 | 2.1800 | 3.1047 | 3.7872 | 3.1554 | 2.6239 | 2.5166 | 3.0901 | 2.5447 | | 1.7446 | H13 | 2.7997 | 2.1372 | 1.1004 | 2.1800 | 3.1042 | 3.7873 | 2.6241 | 3.1564 | 2.5164 | 2.5449 | 3.0902 | 1.7446 | |
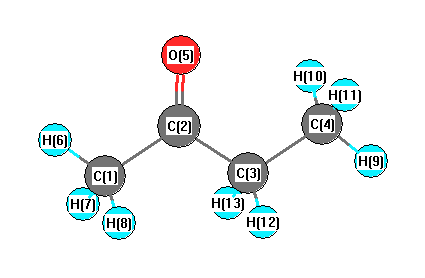 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
116.558 |
|
C1 |
C2 |
O5 |
121.592 |
| C2 |
C1 |
H6 |
109.804 |
|
C2 |
C1 |
H7 |
110.566 |
| C2 |
C1 |
H8 |
110.578 |
|
C2 |
C3 |
C4 |
113.607 |
| C2 |
C3 |
H12 |
107.859 |
|
C2 |
C3 |
H13 |
107.852 |
| C3 |
C2 |
O5 |
121.850 |
|
C3 |
C4 |
H9 |
110.719 |
| C3 |
C4 |
H10 |
110.911 |
|
C3 |
C4 |
H11 |
110.911 |
| C4 |
C3 |
H12 |
111.098 |
|
C4 |
C3 |
H13 |
111.099 |
| H6 |
C1 |
H7 |
109.493 |
|
H6 |
C1 |
H8 |
109.509 |
| H7 |
C1 |
H8 |
106.839 |
|
H9 |
C4 |
H10 |
108.479 |
| H9 |
C4 |
H11 |
108.470 |
|
H10 |
C4 |
H11 |
107.233 |
| H12 |
C3 |
H13 |
104.883 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYPultrafine/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.528 |
|
|
|
| 2 |
C |
0.455 |
|
|
|
| 3 |
C |
-0.347 |
|
|
|
| 4 |
C |
-0.433 |
|
|
|
| 5 |
O |
-0.434 |
|
|
|
| 6 |
H |
0.180 |
|
|
|
| 7 |
H |
0.166 |
|
|
|
| 8 |
H |
0.166 |
|
|
|
| 9 |
H |
0.139 |
|
|
|
| 10 |
H |
0.161 |
|
|
|
| 11 |
H |
0.161 |
|
|
|
| 12 |
H |
0.157 |
|
|
|
| 13 |
H |
0.157 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.227 |
-2.681 |
-0.000 |
2.691 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.875 |
1.109 |
-0.000 |
| y |
1.109 |
-34.650 |
0.000 |
| z |
-0.000 |
0.000 |
-30.273 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.586 |
1.109 |
-0.000 |
| y |
1.109 |
-4.576 |
0.000 |
| z |
-0.000 |
0.000 |
1.990 |
|
| Polar |
|---|
| 3z2-r2 | 3.980 |
|---|
| x2-y2 | 4.774 |
|---|
| xy | 1.109 |
|---|
| xz | -0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.369 |
0.134 |
0.000 |
| y |
0.134 |
6.961 |
0.000 |
| z |
0.000 |
0.000 |
5.322 |
<r2> (average value of r
2) Å
2
| <r2> |
137.593 |
| (<r2>)1/2 |
11.730 |