Vibrational Frequencies calculated at B3LYPultrafine/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3670 |
3551 |
67.78 |
|
|
|
| 2 |
A' |
3255 |
3149 |
1.96 |
|
|
|
| 3 |
A' |
3236 |
3131 |
1.36 |
|
|
|
| 4 |
A' |
3189 |
3086 |
15.99 |
|
|
|
| 5 |
A' |
3178 |
3075 |
27.70 |
|
|
|
| 6 |
A' |
3167 |
3064 |
2.80 |
|
|
|
| 7 |
A' |
3161 |
3058 |
1.75 |
|
|
|
| 8 |
A' |
1655 |
1602 |
3.02 |
|
|
|
| 9 |
A' |
1615 |
1562 |
0.88 |
|
|
|
| 10 |
A' |
1548 |
1498 |
6.48 |
|
|
|
| 11 |
A' |
1525 |
1476 |
2.05 |
|
|
|
| 12 |
A' |
1481 |
1433 |
21.05 |
|
|
|
| 13 |
A' |
1441 |
1394 |
16.70 |
|
|
|
| 14 |
A' |
1386 |
1341 |
6.78 |
|
|
|
| 15 |
A' |
1362 |
1318 |
22.27 |
|
|
|
| 16 |
A' |
1296 |
1254 |
20.40 |
|
|
|
| 17 |
A' |
1269 |
1228 |
10.01 |
|
|
|
| 18 |
A' |
1224 |
1184 |
3.12 |
|
|
|
| 19 |
A' |
1174 |
1136 |
1.34 |
|
|
|
| 20 |
A' |
1142 |
1105 |
0.15 |
|
|
|
| 21 |
A' |
1108 |
1072 |
23.26 |
|
|
|
| 22 |
A' |
1087 |
1052 |
8.87 |
|
|
|
| 23 |
A' |
1033 |
1000 |
6.85 |
|
|
|
| 24 |
A' |
912 |
882 |
6.56 |
|
|
|
| 25 |
A' |
889 |
860 |
0.36 |
|
|
|
| 26 |
A' |
776 |
750 |
2.26 |
|
|
|
| 27 |
A' |
619 |
598 |
1.15 |
|
|
|
| 28 |
A' |
552 |
534 |
0.09 |
|
|
|
| 29 |
A' |
404 |
391 |
4.31 |
|
|
|
| 30 |
A" |
991 |
958 |
0.02 |
|
|
|
| 31 |
A" |
951 |
920 |
2.08 |
|
|
|
| 32 |
A" |
880 |
852 |
2.00 |
|
|
|
| 33 |
A" |
865 |
837 |
0.36 |
|
|
|
| 34 |
A" |
788 |
763 |
13.05 |
|
|
|
| 35 |
A" |
759 |
734 |
93.50 |
|
|
|
| 36 |
A" |
733 |
709 |
28.12 |
|
|
|
| 37 |
A" |
620 |
600 |
6.94 |
|
|
|
| 38 |
A" |
589 |
570 |
0.62 |
|
|
|
| 39 |
A" |
435 |
421 |
1.32 |
|
|
|
| 40 |
A" |
408 |
395 |
68.03 |
|
|
|
| 41 |
A" |
246 |
238 |
0.14 |
|
|
|
| 42 |
A" |
214 |
207 |
10.80 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28416.4 cm
-1
Scaled (by 0.9675) Zero Point Vibrational Energy (zpe) 27492.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
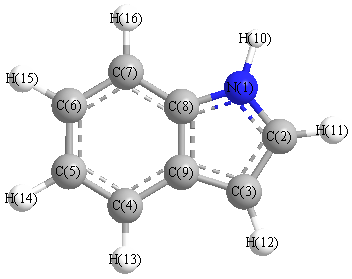
Charges from optimized geometry at B3LYPultrafine/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
0.030 |
|
|
|
| 2 |
C |
-0.633 |
|
|
|
| 3 |
C |
-0.916 |
|
|
|
| 4 |
C |
-0.886 |
|
|
|
| 5 |
C |
-0.216 |
|
|
|
| 6 |
C |
-0.345 |
|
|
|
| 7 |
C |
-0.698 |
|
|
|
| 8 |
C |
0.079 |
|
|
|
| 9 |
C |
1.386 |
|
|
|
| 10 |
H |
-0.059 |
|
|
|
| 11 |
H |
0.627 |
|
|
|
| 12 |
H |
0.288 |
|
|
|
| 13 |
H |
0.288 |
|
|
|
| 14 |
H |
0.383 |
|
|
|
| 15 |
H |
0.361 |
|
|
|
| 16 |
H |
0.311 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.884 |
1.916 |
0.000 |
2.110 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-48.090 |
-1.989 |
0.000 |
| y |
-1.989 |
-43.361 |
0.000 |
| z |
0.000 |
0.000 |
-58.069 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.625 |
-1.989 |
0.000 |
| y |
-1.989 |
9.719 |
0.000 |
| z |
0.000 |
0.000 |
-12.344 |
|
| Polar |
|---|
| 3z2-r2 | -24.687 |
|---|
| x2-y2 | -4.729 |
|---|
| xy | -1.989 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
20.389 |
-1.603 |
0.000 |
| y |
-1.603 |
16.423 |
0.000 |
| z |
0.000 |
0.000 |
8.848 |
<r2> (average value of r
2) Å
2
| <r2> |
281.711 |
| (<r2>)1/2 |
16.784 |