Jump to
S1C2
S1C3
Energy calculated at B3LYPultrafine/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -255.197823 |
| Energy at 298.15K | -255.197240 |
| HF Energy | -255.197823 |
| Nuclear repulsion energy | 46.700244 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYPultrafine/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
2215 |
2143 |
0.44 |
|
|
|
| 2 |
Σ |
368 |
357 |
58.82 |
|
|
|
| 3 |
Π |
127 |
123 |
11.21 |
|
|
|
| 3 |
Π |
127 |
123 |
11.21 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 1418.5 cm
-1
Scaled (by 0.9675) Zero Point Vibrational Energy (zpe) 1372.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYPultrafine/aug-cc-pVTZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-0.688 |
| N2 |
0.000 |
0.000 |
-1.850 |
| Na3 |
0.000 |
0.000 |
1.553 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.1612 | 2.2410 |
N2 | 1.1612 | | 3.4023 | Na3 | 2.2410 | 3.4023 | |
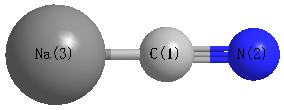 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
0.000 |
|
C1 |
Na3 |
N2 |
0.000 |
| N2 |
C1 |
Na3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYPultrafine/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.313 |
|
|
|
| 2 |
N |
-0.261 |
|
|
|
| 3 |
Na |
0.574 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
10.776 |
10.776 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-17.383 |
0.000 |
0.000 |
| y |
0.000 |
-17.383 |
0.000 |
| z |
0.000 |
0.000 |
-14.114 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.634 |
0.000 |
0.000 |
| y |
0.000 |
-1.634 |
0.000 |
| z |
0.000 |
0.000 |
3.269 |
|
| Polar |
|---|
| 3z2-r2 | 6.538 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.585 |
0.000 |
0.000 |
| y |
0.000 |
3.585 |
0.000 |
| z |
0.000 |
0.000 |
6.329 |
<r2> (average value of r
2) Å
2
| <r2> |
63.486 |
| (<r2>)1/2 |
7.968 |
Jump to
S1C1
S1C3
Energy calculated at B3LYPultrafine/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -255.201043 |
| Energy at 298.15K | -255.200794 |
| HF Energy | -255.201043 |
| Nuclear repulsion energy | 51.871869 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at B3LYPultrafine/aug-cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.107 |
0.684 |
0.000 |
| N2 |
0.000 |
1.072 |
0.000 |
| Na3 |
-0.604 |
-1.055 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.1730 | 2.4391 |
N2 | 1.1730 | | 2.2106 | Na3 | 2.4391 | 2.2106 | |
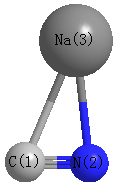 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
86.532 |
|
C1 |
Na3 |
N2 |
28.687 |
| N2 |
C1 |
Na3 |
64.780 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYPultrafine/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.307 |
|
|
|
| 2 |
N |
-0.317 |
|
|
|
| 3 |
Na |
0.624 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-4.578 |
-7.394 |
0.000 |
8.697 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-19.802 |
3.362 |
0.000 |
| y |
3.362 |
-14.325 |
0.000 |
| z |
0.000 |
0.000 |
-17.358 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.961 |
3.362 |
0.000 |
| y |
3.362 |
4.255 |
0.000 |
| z |
0.000 |
0.000 |
-0.294 |
|
| Polar |
|---|
| 3z2-r2 | -0.588 |
|---|
| x2-y2 | -5.477 |
|---|
| xy | 3.362 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.632 |
0.049 |
0.000 |
| y |
0.049 |
4.342 |
0.000 |
| z |
0.000 |
0.000 |
3.530 |
<r2> (average value of r
2) Å
2
| <r2> |
44.755 |
| (<r2>)1/2 |
6.690 |
Jump to
S1C1
S1C2
Energy calculated at B3LYPultrafine/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -255.198349 |
| Energy at 298.15K | -255.197568 |
| HF Energy | -255.198349 |
| Nuclear repulsion energy | 48.863111 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYPultrafine/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
2146 |
2076 |
114.96 |
|
|
|
| 2 |
Σ |
410 |
396 |
67.38 |
|
|
|
| 3 |
Π |
60 |
58 |
4.71 |
|
|
|
| 3 |
Π |
60 |
58 |
4.71 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 1337.5 cm
-1
Scaled (by 0.9675) Zero Point Vibrational Energy (zpe) 1294.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYPultrafine/aug-cc-pVTZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.848 |
| N2 |
0.000 |
0.000 |
-0.677 |
| Na3 |
0.000 |
0.000 |
1.439 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.1710 | 3.2869 |
N2 | 1.1710 | | 2.1159 | Na3 | 3.2869 | 2.1159 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
180.000 |
|
C1 |
Na3 |
N2 |
0.000 |
| N2 |
C1 |
Na3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYPultrafine/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.269 |
|
|
|
| 2 |
N |
-0.462 |
|
|
|
| 3 |
Na |
0.731 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
10.870 |
10.870 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-17.370 |
0.000 |
0.000 |
| y |
0.000 |
-17.370 |
0.000 |
| z |
0.000 |
0.000 |
-16.392 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.489 |
0.000 |
0.000 |
| y |
0.000 |
-0.489 |
0.000 |
| z |
0.000 |
0.000 |
0.978 |
|
| Polar |
|---|
| 3z2-r2 | 1.956 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.601 |
0.000 |
0.000 |
| y |
0.000 |
3.601 |
0.000 |
| z |
0.000 |
0.000 |
6.214 |
<r2> (average value of r
2) Å
2
| <r2> |
57.120 |
| (<r2>)1/2 |
7.558 |