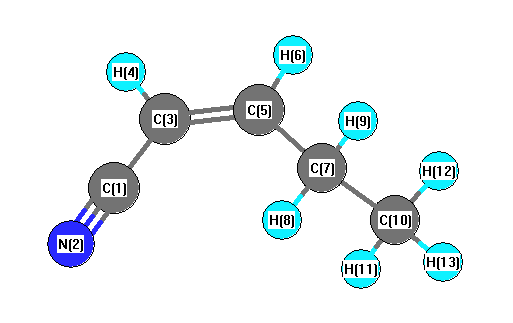
Vibrational Frequencies calculated at B3LYPultrafine/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3176 |
3073 |
2.66 |
|
|
|
| 2 |
A |
3140 |
3038 |
8.46 |
|
|
|
| 3 |
A |
3102 |
3001 |
22.73 |
|
|
|
| 4 |
A |
3096 |
2995 |
24.73 |
|
|
|
| 5 |
A |
3071 |
2971 |
1.06 |
|
|
|
| 6 |
A |
3031 |
2933 |
27.24 |
|
|
|
| 7 |
A |
3009 |
2911 |
11.78 |
|
|
|
| 8 |
A |
2325 |
2249 |
22.93 |
|
|
|
| 9 |
A |
1678 |
1624 |
18.72 |
|
|
|
| 10 |
A |
1507 |
1458 |
7.18 |
|
|
|
| 11 |
A |
1500 |
1451 |
6.42 |
|
|
|
| 12 |
A |
1484 |
1436 |
4.72 |
|
|
|
| 13 |
A |
1431 |
1385 |
0.83 |
|
|
|
| 14 |
A |
1410 |
1365 |
0.82 |
|
|
|
| 15 |
A |
1346 |
1302 |
2.53 |
|
|
|
| 16 |
A |
1295 |
1253 |
0.14 |
|
|
|
| 17 |
A |
1260 |
1219 |
0.75 |
|
|
|
| 18 |
A |
1146 |
1109 |
0.40 |
|
|
|
| 19 |
A |
1094 |
1058 |
3.17 |
|
|
|
| 20 |
A |
1021 |
987 |
5.03 |
|
|
|
| 21 |
A |
1007 |
974 |
0.83 |
|
|
|
| 22 |
A |
963 |
932 |
4.12 |
|
|
|
| 23 |
A |
863 |
835 |
3.32 |
|
|
|
| 24 |
A |
809 |
783 |
7.26 |
|
|
|
| 25 |
A |
765 |
741 |
33.52 |
|
|
|
| 26 |
A |
675 |
653 |
1.56 |
|
|
|
| 27 |
A |
574 |
555 |
0.99 |
|
|
|
| 28 |
A |
400 |
387 |
0.42 |
|
|
|
| 29 |
A |
366 |
354 |
0.03 |
|
|
|
| 30 |
A |
242 |
234 |
0.99 |
|
|
|
| 31 |
A |
216 |
209 |
2.77 |
|
|
|
| 32 |
A |
145 |
140 |
5.07 |
|
|
|
| 33 |
A |
58 |
56 |
1.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23601.3 cm
-1
Scaled (by 0.9675) Zero Point Vibrational Energy (zpe) 22834.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYPultrafine/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.663 |
|
|
|
| 2 |
N |
-0.683 |
|
|
|
| 3 |
C |
-0.122 |
|
|
|
| 4 |
H |
0.294 |
|
|
|
| 5 |
C |
-0.485 |
|
|
|
| 6 |
H |
0.291 |
|
|
|
| 7 |
C |
-0.106 |
|
|
|
| 8 |
H |
0.097 |
|
|
|
| 9 |
H |
0.160 |
|
|
|
| 10 |
C |
-0.714 |
|
|
|
| 11 |
H |
0.227 |
|
|
|
| 12 |
H |
0.206 |
|
|
|
| 13 |
H |
0.173 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.547 |
2.516 |
0.081 |
4.349 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-43.646 |
-5.857 |
1.041 |
| y |
-5.857 |
-36.700 |
-0.660 |
| z |
1.041 |
-0.660 |
-37.503 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.545 |
-5.857 |
1.041 |
| y |
-5.857 |
3.875 |
-0.660 |
| z |
1.041 |
-0.660 |
2.670 |
|
| Polar |
|---|
| 3z2-r2 | 5.340 |
|---|
| x2-y2 | -6.947 |
|---|
| xy | -5.857 |
|---|
| xz | 1.041 |
|---|
| yz | -0.660 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.416 |
0.942 |
0.336 |
| y |
0.942 |
9.581 |
-0.154 |
| z |
0.336 |
-0.154 |
7.581 |
<r2> (average value of r
2) Å
2
| <r2> |
201.981 |
| (<r2>)1/2 |
14.212 |