Jump to
S1C2
S1C3
Energy calculated at MP3=FULL/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.069460 |
| Energy at 298.15K | -100.068441 |
| HF Energy | -99.780208 |
| Nuclear repulsion energy | 27.392059 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP3=FULL/cc-pVDZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
-2.096 |
| C2 |
0.000 |
0.000 |
-0.148 |
| N3 |
0.000 |
0.000 |
1.025 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 1.9478 | 3.1212 |
C2 | 1.9478 | | 1.1734 | N3 | 3.1212 | 1.1734 | |
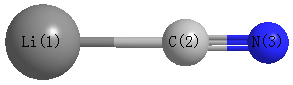 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
180.000 |
|
Li1 |
N3 |
C2 |
0.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at MP3=FULL/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.075681 |
| Energy at 298.15K | -100.074858 |
| HF Energy | -99.789766 |
| Nuclear repulsion energy | 28.872310 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP3=FULL/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
1.451 |
-0.595 |
0.000 |
| C2 |
-0.725 |
-0.368 |
0.000 |
| N3 |
0.000 |
0.571 |
0.000 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.1880 | 1.8608 |
C2 | 2.1880 | | 1.1864 | N3 | 1.8608 | 1.1864 | |
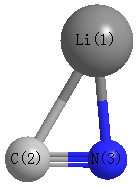 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
58.245 |
|
Li1 |
N3 |
C2 |
88.924 |
| C2 |
Li1 |
N3 |
32.830 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP3=FULL/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.074569 |
| Energy at 298.15K | -100.073158 |
| HF Energy | -99.792141 |
| Nuclear repulsion energy | 28.093147 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP3=FULL/cc-pVDZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
1.910 |
| C2 |
0.000 |
0.000 |
-1.079 |
| N3 |
0.000 |
0.000 |
0.107 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.9890 | 1.8032 |
C2 | 2.9890 | | 1.1858 | N3 | 1.8032 | 1.1858 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
0.000 |
|
Li1 |
N3 |
C2 |
180.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability