Vibrational Frequencies calculated at TPSSh/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3093 |
2995 |
27.88 |
|
|
|
| 2 |
A' |
3089 |
2991 |
40.37 |
|
|
|
| 3 |
A' |
3028 |
2932 |
29.95 |
|
|
|
| 4 |
A' |
3028 |
2932 |
35.56 |
|
|
|
| 5 |
A' |
3024 |
2928 |
34.87 |
|
|
|
| 6 |
A' |
3019 |
2924 |
32.75 |
|
|
|
| 7 |
A' |
3014 |
2918 |
14.39 |
|
|
|
| 8 |
A' |
1518 |
1469 |
3.72 |
|
|
|
| 9 |
A' |
1514 |
1466 |
1.95 |
|
|
|
| 10 |
A' |
1503 |
1455 |
0.38 |
|
|
|
| 11 |
A' |
1495 |
1447 |
0.93 |
|
|
|
| 12 |
A' |
1487 |
1440 |
7.51 |
|
|
|
| 13 |
A' |
1417 |
1372 |
1.12 |
|
|
|
| 14 |
A' |
1413 |
1369 |
1.91 |
|
|
|
| 15 |
A' |
1367 |
1324 |
0.06 |
|
|
|
| 16 |
A' |
1295 |
1254 |
15.85 |
|
|
|
| 17 |
A' |
1245 |
1206 |
37.00 |
|
|
|
| 18 |
A' |
1116 |
1081 |
2.20 |
|
|
|
| 19 |
A' |
1060 |
1027 |
3.54 |
|
|
|
| 20 |
A' |
1042 |
1009 |
0.20 |
|
|
|
| 21 |
A' |
986 |
955 |
4.35 |
|
|
|
| 22 |
A' |
901 |
872 |
2.08 |
|
|
|
| 23 |
A' |
756 |
732 |
2.28 |
|
|
|
| 24 |
A' |
683 |
661 |
1.14 |
|
|
|
| 25 |
A' |
368 |
356 |
0.30 |
|
|
|
| 26 |
A' |
292 |
283 |
0.49 |
|
|
|
| 27 |
A' |
258 |
250 |
0.86 |
|
|
|
| 28 |
A' |
98 |
95 |
0.27 |
|
|
|
| 29 |
A" |
3104 |
3006 |
30.62 |
|
|
|
| 30 |
A" |
3088 |
2990 |
69.35 |
|
|
|
| 31 |
A" |
3067 |
2970 |
0.39 |
|
|
|
| 32 |
A" |
3062 |
2965 |
15.89 |
|
|
|
| 33 |
A" |
3045 |
2949 |
1.61 |
|
|
|
| 34 |
A" |
1510 |
1463 |
6.27 |
|
|
|
| 35 |
A" |
1502 |
1454 |
7.36 |
|
|
|
| 36 |
A" |
1322 |
1280 |
0.27 |
|
|
|
| 37 |
A" |
1266 |
1226 |
0.00 |
|
|
|
| 38 |
A" |
1241 |
1202 |
0.05 |
|
|
|
| 39 |
A" |
1058 |
1024 |
0.78 |
|
|
|
| 40 |
A" |
1032 |
999 |
0.09 |
|
|
|
| 41 |
A" |
860 |
833 |
0.02 |
|
|
|
| 42 |
A" |
782 |
757 |
3.51 |
|
|
|
| 43 |
A" |
739 |
716 |
2.52 |
|
|
|
| 44 |
A" |
238 |
230 |
0.03 |
|
|
|
| 45 |
A" |
233 |
225 |
0.12 |
|
|
|
| 46 |
A" |
111 |
108 |
0.89 |
|
|
|
| 47 |
A" |
67 |
65 |
0.53 |
|
|
|
| 48 |
A" |
40 |
39 |
0.26 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35237.0 cm
-1
Scaled (by 0.9683) Zero Point Vibrational Energy (zpe) 34120.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
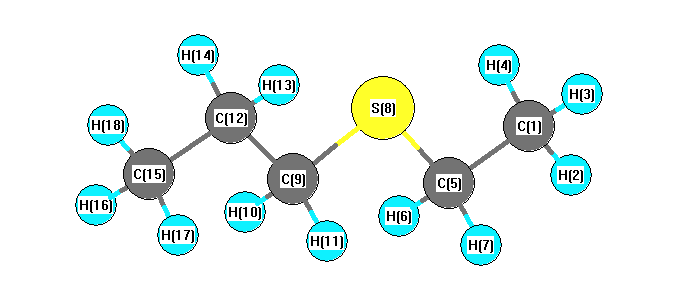
Charges from optimized geometry at TPSSh/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.229 |
|
|
|
| 2 |
H |
0.075 |
|
|
|
| 3 |
H |
0.083 |
|
|
|
| 4 |
H |
0.083 |
|
|
|
| 5 |
C |
-0.108 |
|
|
|
| 6 |
H |
0.078 |
|
|
|
| 7 |
H |
0.078 |
|
|
|
| 8 |
S |
-0.139 |
|
|
|
| 9 |
C |
-0.088 |
|
|
|
| 10 |
H |
0.073 |
|
|
|
| 11 |
H |
0.073 |
|
|
|
| 12 |
C |
-0.116 |
|
|
|
| 13 |
H |
0.075 |
|
|
|
| 14 |
H |
0.075 |
|
|
|
| 15 |
C |
-0.231 |
|
|
|
| 16 |
H |
0.070 |
|
|
|
| 17 |
H |
0.070 |
|
|
|
| 18 |
H |
0.078 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.920 |
-1.251 |
0.000 |
1.553 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.906 |
2.782 |
0.000 |
| y |
2.782 |
13.716 |
0.000 |
| z |
0.000 |
0.000 |
9.567 |
<r2> (average value of r
2) Å
2
| <r2> |
354.412 |
| (<r2>)1/2 |
18.826 |