Vibrational Frequencies calculated at TPSSh/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3109 |
3010 |
30.31 |
|
|
|
| 2 |
A' |
3093 |
2995 |
26.93 |
|
|
|
| 3 |
A' |
3082 |
2984 |
65.85 |
|
|
|
| 4 |
A' |
3038 |
2942 |
20.32 |
|
|
|
| 5 |
A' |
3035 |
2939 |
7.62 |
|
|
|
| 6 |
A' |
3028 |
2932 |
32.89 |
|
|
|
| 7 |
A' |
3021 |
2925 |
38.89 |
|
|
|
| 8 |
A' |
1517 |
1469 |
3.16 |
|
|
|
| 9 |
A' |
1514 |
1466 |
3.34 |
|
|
|
| 10 |
A' |
1509 |
1461 |
11.86 |
|
|
|
| 11 |
A' |
1495 |
1447 |
1.94 |
|
|
|
| 12 |
A' |
1421 |
1376 |
2.66 |
|
|
|
| 13 |
A' |
1415 |
1370 |
1.12 |
|
|
|
| 14 |
A' |
1288 |
1247 |
24.40 |
|
|
|
| 15 |
A' |
1285 |
1245 |
13.67 |
|
|
|
| 16 |
A' |
1184 |
1147 |
9.12 |
|
|
|
| 17 |
A' |
1093 |
1059 |
18.28 |
|
|
|
| 18 |
A' |
1043 |
1010 |
0.47 |
|
|
|
| 19 |
A' |
985 |
954 |
3.48 |
|
|
|
| 20 |
A' |
887 |
859 |
1.35 |
|
|
|
| 21 |
A' |
686 |
664 |
0.74 |
|
|
|
| 22 |
A' |
605 |
586 |
2.12 |
|
|
|
| 23 |
A' |
454 |
439 |
1.35 |
|
|
|
| 24 |
A' |
351 |
340 |
0.11 |
|
|
|
| 25 |
A' |
275 |
266 |
0.78 |
|
|
|
| 26 |
A' |
242 |
235 |
0.13 |
|
|
|
| 27 |
A' |
147 |
142 |
0.23 |
|
|
|
| 28 |
A" |
3107 |
3009 |
8.66 |
|
|
|
| 29 |
A" |
3106 |
3008 |
43.97 |
|
|
|
| 30 |
A" |
3079 |
2981 |
4.11 |
|
|
|
| 31 |
A" |
3075 |
2977 |
0.04 |
|
|
|
| 32 |
A" |
3017 |
2921 |
19.82 |
|
|
|
| 33 |
A" |
1503 |
1456 |
1.48 |
|
|
|
| 34 |
A" |
1501 |
1453 |
6.86 |
|
|
|
| 35 |
A" |
1493 |
1446 |
1.07 |
|
|
|
| 36 |
A" |
1402 |
1357 |
5.73 |
|
|
|
| 37 |
A" |
1335 |
1292 |
1.14 |
|
|
|
| 38 |
A" |
1268 |
1228 |
0.03 |
|
|
|
| 39 |
A" |
1123 |
1088 |
1.45 |
|
|
|
| 40 |
A" |
1036 |
1003 |
0.08 |
|
|
|
| 41 |
A" |
950 |
920 |
0.39 |
|
|
|
| 42 |
A" |
925 |
896 |
0.62 |
|
|
|
| 43 |
A" |
787 |
762 |
2.54 |
|
|
|
| 44 |
A" |
308 |
299 |
1.72 |
|
|
|
| 45 |
A" |
235 |
228 |
0.07 |
|
|
|
| 46 |
A" |
225 |
218 |
0.01 |
|
|
|
| 47 |
A" |
64 |
62 |
1.01 |
|
|
|
| 48 |
A" |
53 |
52 |
0.90 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35197.0 cm
-1
Scaled (by 0.9683) Zero Point Vibrational Energy (zpe) 34081.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
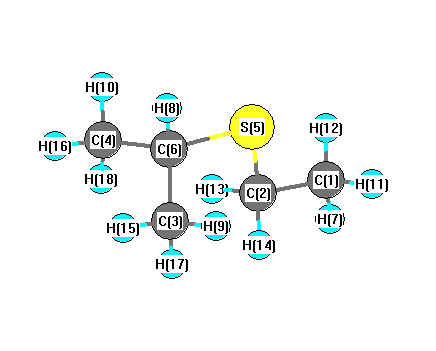
Charges from optimized geometry at TPSSh/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.225 |
|
|
|
| 2 |
C |
-0.111 |
|
|
|
| 3 |
C |
-0.210 |
|
|
|
| 4 |
C |
-0.210 |
|
|
|
| 5 |
S |
-0.140 |
|
|
|
| 6 |
C |
-0.037 |
|
|
|
| 7 |
H |
0.075 |
|
|
|
| 8 |
H |
0.075 |
|
|
|
| 9 |
H |
0.086 |
|
|
|
| 10 |
H |
0.086 |
|
|
|
| 11 |
H |
0.082 |
|
|
|
| 12 |
H |
0.082 |
|
|
|
| 13 |
H |
0.082 |
|
|
|
| 14 |
H |
0.082 |
|
|
|
| 15 |
H |
0.069 |
|
|
|
| 16 |
H |
0.069 |
|
|
|
| 17 |
H |
0.072 |
|
|
|
| 18 |
H |
0.072 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.129 |
-1.619 |
0.000 |
1.624 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
14.247 |
1.518 |
0.000 |
| y |
1.518 |
11.694 |
0.000 |
| z |
0.000 |
0.000 |
10.714 |
<r2> (average value of r
2) Å
2
| <r2> |
266.906 |
| (<r2>)1/2 |
16.337 |