Jump to
S1C2
Energy calculated at B2PLYP=FULLultrafine/6-31G*
| | hartrees |
|---|
| Energy at 0K | -302.807626 |
| Energy at 298.15K | |
| HF Energy | -302.540053 |
| Nuclear repulsion energy | 158.785362 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULLultrafine/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3080 |
2922 |
94.12 |
108.96 |
0.28 |
0.44 |
| 2 |
A1 |
1914 |
1816 |
25.34 |
25.29 |
0.18 |
0.31 |
| 3 |
A1 |
1482 |
1406 |
0.05 |
3.25 |
0.32 |
0.48 |
| 4 |
A1 |
1148 |
1089 |
111.46 |
5.41 |
0.17 |
0.29 |
| 5 |
A1 |
537 |
509 |
0.65 |
10.09 |
0.35 |
0.52 |
| 6 |
A1 |
280 |
265 |
12.92 |
0.77 |
0.51 |
0.68 |
| 7 |
A2 |
1031 |
978 |
0.00 |
1.76 |
0.75 |
0.86 |
| 8 |
A2 |
172 |
163 |
0.00 |
0.43 |
0.75 |
0.86 |
| 9 |
B1 |
1037 |
984 |
1.06 |
3.65 |
0.75 |
0.86 |
| 10 |
B1 |
129 |
122 |
3.90 |
0.09 |
0.75 |
0.86 |
| 11 |
B2 |
3058 |
2901 |
0.02 |
2.03 |
0.75 |
0.86 |
| 12 |
B2 |
1836 |
1742 |
601.22 |
2.88 |
0.75 |
0.86 |
| 13 |
B2 |
1426 |
1353 |
9.52 |
10.27 |
0.75 |
0.86 |
| 14 |
B2 |
1095 |
1039 |
623.37 |
2.35 |
0.75 |
0.86 |
| 15 |
B2 |
705 |
669 |
38.74 |
0.00 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 9464.7 cm
-1
Scaled (by 0.9487) Zero Point Vibrational Energy (zpe) 8979.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULLultrafine/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.387 |
| C2 |
0.000 |
1.182 |
-0.323 |
| C3 |
0.000 |
-1.182 |
-0.323 |
| O4 |
0.000 |
2.245 |
0.226 |
| O5 |
0.000 |
-2.245 |
0.226 |
| H6 |
0.000 |
1.036 |
-1.414 |
| H7 |
0.000 |
-1.036 |
-1.414 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
O4 |
O5 |
H6 |
H7 |
| O1 | | 1.3790 | 1.3790 | 2.2506 | 2.2506 | 2.0779 | 2.0779 |
C2 | 1.3790 | | 2.3640 | 1.1961 | 3.4704 | 1.1003 | 2.4720 | C3 | 1.3790 | 2.3640 | | 3.4704 | 1.1961 | 2.4720 | 1.1003 | O4 | 2.2506 | 1.1961 | 3.4704 | | 4.4895 | 2.0366 | 3.6679 | O5 | 2.2506 | 3.4704 | 1.1961 | 4.4895 | | 3.6679 | 2.0366 | H6 | 2.0779 | 1.1003 | 2.4720 | 2.0366 | 3.6679 | | 2.0728 | H7 | 2.0779 | 2.4720 | 1.1003 | 3.6679 | 2.0366 | 2.0728 | |
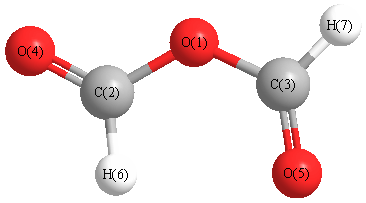 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
O4 |
121.685 |
|
O1 |
C2 |
H6 |
113.403 |
| O1 |
C3 |
O5 |
121.685 |
|
C2 |
O1 |
C3 |
117.988 |
| O4 |
C2 |
H6 |
124.912 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP=FULLultrafine/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.393 |
|
|
|
| 2 |
C |
0.406 |
|
|
|
| 3 |
C |
0.406 |
|
|
|
| 4 |
O |
-0.357 |
|
|
|
| 5 |
O |
-0.357 |
|
|
|
| 6 |
H |
0.147 |
|
|
|
| 7 |
H |
0.147 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.268 |
3.268 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-26.125 |
0.000 |
0.000 |
| y |
0.000 |
-38.104 |
0.000 |
| z |
0.000 |
0.000 |
-25.865 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.859 |
0.000 |
0.000 |
| y |
0.000 |
-12.109 |
0.000 |
| z |
0.000 |
0.000 |
6.250 |
|
| Polar |
|---|
| 3z2-r2 | 12.499 |
|---|
| x2-y2 | 11.979 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.156 |
0.000 |
0.000 |
| y |
0.000 |
6.947 |
0.000 |
| z |
0.000 |
0.000 |
3.775 |
<r2> (average value of r
2) Å
2
| <r2> |
125.558 |
| (<r2>)1/2 |
11.205 |
Jump to
S1C1
Energy calculated at B2PLYP=FULLultrafine/6-31G*
| | hartrees |
|---|
| Energy at 0K | -302.813685 |
| Energy at 298.15K | |
| HF Energy | -302.546743 |
| Nuclear repulsion energy | 161.291849 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULLultrafine/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3165 |
3003 |
15.33 |
33.88 |
0.34 |
0.51 |
| 2 |
A' |
3142 |
2981 |
39.22 |
123.34 |
0.28 |
0.44 |
| 3 |
A' |
1873 |
1777 |
126.56 |
18.93 |
0.20 |
0.34 |
| 4 |
A' |
1824 |
1730 |
367.56 |
2.63 |
0.39 |
0.56 |
| 5 |
A' |
1434 |
1361 |
3.47 |
5.95 |
0.56 |
0.71 |
| 6 |
A' |
1413 |
1340 |
0.71 |
7.44 |
0.72 |
0.84 |
| 7 |
A' |
1144 |
1085 |
644.38 |
2.73 |
0.22 |
0.37 |
| 8 |
A' |
1030 |
977 |
179.46 |
5.58 |
0.11 |
0.19 |
| 9 |
A' |
788 |
747 |
1.86 |
5.76 |
0.57 |
0.72 |
| 10 |
A' |
538 |
511 |
4.78 |
4.26 |
0.42 |
0.60 |
| 11 |
A' |
260 |
246 |
9.73 |
1.68 |
0.44 |
0.61 |
| 12 |
A" |
1052 |
998 |
0.09 |
4.92 |
0.75 |
0.86 |
| 13 |
A" |
1037 |
984 |
0.42 |
1.05 |
0.75 |
0.86 |
| 14 |
A" |
234 |
222 |
15.08 |
1.40 |
0.75 |
0.86 |
| 15 |
A" |
104 |
99 |
33.76 |
0.96 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 9518.4 cm
-1
Scaled (by 0.9487) Zero Point Vibrational Energy (zpe) 9030.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULLultrafine/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.724 |
0.000 |
| C2 |
0.982 |
-0.263 |
0.000 |
| C3 |
-1.317 |
0.328 |
0.000 |
| O4 |
2.140 |
0.039 |
0.000 |
| O5 |
-1.717 |
-0.806 |
0.000 |
| H6 |
0.572 |
-1.277 |
0.000 |
| H7 |
-1.942 |
1.227 |
0.000 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
O4 |
O5 |
H6 |
H7 |
| O1 | | 1.3927 | 1.3751 | 2.2466 | 2.3000 | 2.0810 | 2.0066 |
C2 | 1.3927 | | 2.3740 | 1.1967 | 2.7535 | 1.0933 | 3.2826 | C3 | 1.3751 | 2.3740 | | 3.4688 | 1.2027 | 2.4788 | 1.0954 | O4 | 2.2466 | 1.1967 | 3.4688 | | 3.9488 | 2.0472 | 4.2516 | O5 | 2.3000 | 2.7535 | 1.2027 | 3.9488 | | 2.3373 | 2.0457 | H6 | 2.0810 | 1.0933 | 2.4788 | 2.0472 | 2.3373 | | 3.5488 | H7 | 2.0066 | 3.2826 | 1.0954 | 4.2516 | 2.0457 | 3.5488 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
O4 |
120.182 |
|
O1 |
C2 |
H6 |
113.118 |
| O1 |
C3 |
O5 |
126.175 |
|
C2 |
O1 |
C3 |
118.120 |
| O4 |
C2 |
H6 |
126.699 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP=FULLultrafine/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.419 |
|
|
|
| 2 |
C |
0.393 |
|
|
|
| 3 |
C |
0.397 |
|
|
|
| 4 |
O |
-0.359 |
|
|
|
| 5 |
O |
-0.379 |
|
|
|
| 6 |
H |
0.190 |
|
|
|
| 7 |
H |
0.177 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.685 |
0.444 |
0.000 |
1.743 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.458 |
-5.302 |
0.001 |
| y |
-5.302 |
-27.469 |
0.000 |
| z |
0.001 |
0.000 |
-26.103 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.672 |
-5.302 |
0.001 |
| y |
-5.302 |
2.312 |
0.000 |
| z |
0.001 |
0.000 |
4.360 |
|
| Polar |
|---|
| 3z2-r2 | 8.720 |
|---|
| x2-y2 | -5.989 |
|---|
| xy | -5.302 |
|---|
| xz | 0.001 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.188 |
0.150 |
-0.000 |
| y |
0.150 |
4.088 |
0.000 |
| z |
-0.000 |
0.000 |
2.147 |
<r2> (average value of r
2) Å
2
| <r2> |
112.240 |
| (<r2>)1/2 |
10.594 |