Jump to
S1C2
S1C3
Energy calculated at B2PLYP=FULLultrafine/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -579.779393 |
| Energy at 298.15K | |
| HF Energy | -579.705216 |
| Nuclear repulsion energy | 66.585222 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULLultrafine/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2355 |
2256 |
0.00 |
311.29 |
0.31 |
0.47 |
| 2 |
Σg |
744 |
713 |
0.00 |
71.93 |
0.23 |
0.38 |
| 3 |
Σu |
2352 |
2254 |
1.36 |
0.00 |
0.30 |
0.00 |
| 4 |
Πg |
623i |
597i |
0.00 |
34.68 |
0.75 |
0.86 |
| 4 |
Πg |
623i |
597i |
0.00 |
34.68 |
0.75 |
0.86 |
| 5 |
Πu |
416 |
399 |
2.78 |
0.00 |
0.00 |
0.00 |
| 5 |
Πu |
416 |
399 |
2.78 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2518.0 cm
-1
Scaled (by 0.9582) Zero Point Vibrational Energy (zpe) 2412.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULLultrafine/cc-pVDZ
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
0.996 |
| Si2 |
0.000 |
0.000 |
-0.996 |
| H3 |
0.000 |
0.000 |
2.462 |
| H4 |
0.000 |
0.000 |
-2.462 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 1.9911 | 1.4666 | 3.4578 |
Si2 | 1.9911 | | 3.4578 | 1.4666 | H3 | 1.4666 | 3.4578 | | 4.9244 | H4 | 3.4578 | 1.4666 | 4.9244 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
180.000 |
|
Si2 |
Si1 |
H3 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP=FULLultrafine/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
-0.026 |
|
|
|
| 2 |
Si |
-0.026 |
|
|
|
| 3 |
H |
0.026 |
|
|
|
| 4 |
H |
0.026 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.106 |
0.000 |
0.000 |
| y |
0.000 |
-30.106 |
0.000 |
| z |
0.000 |
0.000 |
-20.362 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.872 |
0.000 |
0.000 |
| y |
0.000 |
-4.872 |
0.000 |
| z |
0.000 |
0.000 |
9.744 |
|
| Polar |
|---|
| 3z2-r2 | 19.489 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.035 |
0.000 |
0.000 |
| y |
0.000 |
5.035 |
0.000 |
| z |
0.000 |
0.000 |
14.025 |
<r2> (average value of r
2) Å
2
| <r2> |
56.653 |
| (<r2>)1/2 |
7.527 |
Jump to
S1C1
S1C3
Energy calculated at B2PLYP=FULLultrafine/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -579.813455 |
| Energy at 298.15K | |
| HF Energy | -579.736613 |
| Nuclear repulsion energy | 63.544495 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULLultrafine/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2195 |
2103 |
0.00 |
431.66 |
0.37 |
0.54 |
| 2 |
Ag |
624 |
598 |
0.00 |
38.57 |
0.74 |
0.85 |
| 3 |
Ag |
570 |
546 |
0.00 |
36.83 |
0.25 |
0.40 |
| 4 |
Au |
408 |
391 |
8.64 |
0.00 |
0.00 |
0.00 |
| 5 |
Bu |
2201 |
2109 |
119.16 |
0.00 |
0.00 |
0.00 |
| 6 |
Bu |
291 |
279 |
37.32 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3143.9 cm
-1
Scaled (by 0.9582) Zero Point Vibrational Energy (zpe) 3012.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULLultrafine/cc-pVDZ
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.060 |
0.000 |
| Si2 |
0.000 |
-1.060 |
0.000 |
| H3 |
1.224 |
1.923 |
0.000 |
| H4 |
-1.224 |
-1.923 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1191 | 1.4983 | 3.2243 |
Si2 | 2.1191 | | 3.2243 | 1.4983 | H3 | 1.4983 | 3.2243 | | 4.5598 | H4 | 3.2243 | 1.4983 | 4.5598 | |
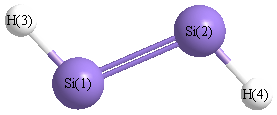 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
125.200 |
|
Si2 |
Si1 |
H3 |
125.200 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP=FULLultrafine/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.024 |
|
|
|
| 2 |
Si |
0.024 |
|
|
|
| 3 |
H |
-0.024 |
|
|
|
| 4 |
H |
-0.024 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-28.787 |
-0.282 |
0.000 |
| y |
-0.282 |
-23.278 |
0.000 |
| z |
0.000 |
0.000 |
-30.809 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.743 |
-0.282 |
0.000 |
| y |
-0.282 |
6.521 |
0.000 |
| z |
0.000 |
0.000 |
-4.777 |
|
| Polar |
|---|
| 3z2-r2 | -9.554 |
|---|
| x2-y2 | -5.509 |
|---|
| xy | -0.282 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.488 |
0.448 |
0.000 |
| y |
0.448 |
14.806 |
0.000 |
| z |
0.000 |
0.000 |
5.638 |
<r2> (average value of r
2) Å
2
| <r2> |
59.085 |
| (<r2>)1/2 |
7.687 |
Jump to
S1C1
S1C2
Energy calculated at B2PLYP=FULLultrafine/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -579.841233 |
| Energy at 298.15K | |
| HF Energy | -579.772313 |
| Nuclear repulsion energy | 64.145481 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP=FULLultrafine/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1619 |
1551 |
5.68 |
81.47 |
0.07 |
0.13 |
| 2 |
A1 |
931 |
892 |
35.82 |
17.02 |
0.43 |
0.60 |
| 3 |
A1 |
515 |
494 |
0.79 |
59.84 |
0.36 |
0.53 |
| 4 |
A2 |
1094 |
1048 |
0.00 |
3.28 |
0.75 |
0.86 |
| 5 |
B1 |
1540 |
1475 |
19.85 |
40.09 |
0.75 |
0.86 |
| 6 |
B2 |
1171 |
1122 |
394.14 |
2.08 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3434.6 cm
-1
Scaled (by 0.9582) Zero Point Vibrational Energy (zpe) 3291.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP=FULLultrafine/cc-pVDZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.119 |
-0.052 |
| Si2 |
0.000 |
-1.119 |
-0.052 |
| H3 |
0.998 |
0.000 |
0.729 |
| H4 |
-0.998 |
0.000 |
0.729 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.2377 | 1.6905 | 1.6905 |
Si2 | 2.2377 | | 1.6905 | 1.6905 | H3 | 1.6905 | 1.6905 | | 1.9966 | H4 | 1.6905 | 1.6905 | 1.9966 | |
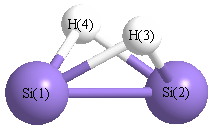 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
48.561 |
|
Si2 |
Si1 |
H3 |
48.561 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP=FULLultrafine/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.029 |
|
|
|
| 2 |
Si |
0.029 |
|
|
|
| 3 |
H |
-0.029 |
|
|
|
| 4 |
H |
-0.029 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.294 |
0.294 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-26.080 |
0.000 |
0.000 |
| y |
0.000 |
-31.849 |
0.000 |
| z |
0.000 |
0.000 |
-28.947 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.318 |
0.000 |
0.000 |
| y |
0.000 |
-4.336 |
0.000 |
| z |
0.000 |
0.000 |
0.018 |
|
| Polar |
|---|
| 3z2-r2 | 0.036 |
|---|
| x2-y2 | 5.769 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.413 |
0.000 |
0.000 |
| y |
0.000 |
12.894 |
0.000 |
| z |
0.000 |
0.000 |
6.030 |
<r2> (average value of r
2) Å
2
| <r2> |
56.267 |
| (<r2>)1/2 |
7.501 |