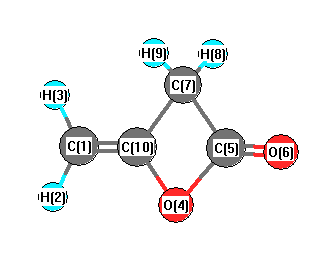
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3282 |
3151 |
1.56 |
|
|
|
| 2 |
A |
3195 |
3068 |
0.16 |
|
|
|
| 3 |
A |
3149 |
3024 |
1.32 |
|
|
|
| 4 |
A |
3098 |
2975 |
2.65 |
|
|
|
| 5 |
A |
1978 |
1899 |
352.76 |
|
|
|
| 6 |
A |
1788 |
1717 |
261.01 |
|
|
|
| 7 |
A |
1474 |
1416 |
9.54 |
|
|
|
| 8 |
A |
1443 |
1385 |
3.55 |
|
|
|
| 9 |
A |
1280 |
1229 |
89.75 |
|
|
|
| 10 |
A |
1223 |
1174 |
0.36 |
|
|
|
| 11 |
A |
1123 |
1078 |
0.70 |
|
|
|
| 12 |
A |
1039 |
997 |
152.39 |
|
|
|
| 13 |
A |
996 |
956 |
5.34 |
|
|
|
| 14 |
A |
995 |
955 |
3.90 |
|
|
|
| 15 |
A |
909 |
873 |
146.92 |
|
|
|
| 16 |
A |
859 |
825 |
55.59 |
|
|
|
| 17 |
A |
822 |
789 |
17.86 |
|
|
|
| 18 |
A |
738 |
709 |
0.00 |
|
|
|
| 19 |
A |
678 |
651 |
0.84 |
|
|
|
| 20 |
A |
526 |
505 |
2.97 |
|
|
|
| 21 |
A |
513 |
493 |
3.54 |
|
|
|
| 22 |
A |
461 |
443 |
4.09 |
|
|
|
| 23 |
A |
317 |
305 |
0.78 |
|
|
|
| 24 |
A |
137 |
132 |
0.97 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16009.4 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 15373.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.462 |
-0.607 |
|
-0.694 |
| 2 |
H |
0.169 |
0.229 |
|
0.255 |
| 3 |
H |
0.159 |
0.205 |
|
0.237 |
| 4 |
O |
-0.456 |
-0.348 |
|
-0.355 |
| 5 |
C |
0.618 |
0.643 |
|
0.678 |
| 6 |
O |
-0.420 |
-0.452 |
|
-0.440 |
| 7 |
C |
-0.494 |
-0.278 |
|
-0.565 |
| 8 |
H |
0.208 |
0.110 |
|
0.199 |
| 9 |
H |
0.208 |
0.110 |
|
0.199 |
| 10 |
C |
0.470 |
0.390 |
|
0.485 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-2.608 |
2.294 |
0.000 |
3.474 |
| CHELPG |
-2.679 |
2.275 |
0.000 |
3.514 |
| AIM |
|
|
|
|
| ESP |
-2.640 |
2.278 |
0.000 |
3.487 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.109 |
1.092 |
0.000 |
| y |
1.092 |
-32.298 |
0.000 |
| z |
0.000 |
0.000 |
-33.416 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.252 |
1.092 |
0.000 |
| y |
1.092 |
3.465 |
0.000 |
| z |
0.000 |
0.000 |
1.787 |
|
| Polar |
|---|
| 3z2-r2 | 3.575 |
|---|
| x2-y2 | -5.811 |
|---|
| xy | 1.092 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.191 |
0.350 |
-0.001 |
| y |
0.350 |
5.762 |
-0.002 |
| z |
-0.001 |
-0.002 |
3.546 |
<r2> (average value of r
2) Å
2
| <r2> |
145.987 |
| (<r2>)1/2 |
12.083 |