Jump to
S1C2
Energy calculated at B3LYP/6-31G*
| | hartrees |
|---|
| Energy at 0K | -230.161080 |
| Energy at 298.15K | -230.168026 |
| Nuclear repulsion energy | 133.522726 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3138 |
3013 |
0.00 |
|
|
|
| 2 |
Ag |
3031 |
2910 |
0.00 |
|
|
|
| 3 |
Ag |
1550 |
1489 |
0.00 |
|
|
|
| 4 |
Ag |
1477 |
1419 |
0.00 |
|
|
|
| 5 |
Ag |
1279 |
1228 |
0.00 |
|
|
|
| 6 |
Ag |
1083 |
1040 |
0.00 |
|
|
|
| 7 |
Ag |
849 |
815 |
0.00 |
|
|
|
| 8 |
Ag |
486 |
467 |
0.00 |
|
|
|
| 9 |
Au |
3092 |
2969 |
104.50 |
|
|
|
| 10 |
Au |
1488 |
1429 |
8.39 |
|
|
|
| 11 |
Au |
1187 |
1140 |
1.47 |
|
|
|
| 12 |
Au |
209 |
201 |
3.51 |
|
|
|
| 13 |
Au |
16 |
15 |
10.73 |
|
|
|
| 14 |
Bg |
3091 |
2968 |
0.00 |
|
|
|
| 15 |
Bg |
1488 |
1428 |
0.00 |
|
|
|
| 16 |
Bg |
1193 |
1146 |
0.00 |
|
|
|
| 17 |
Bg |
266 |
256 |
0.00 |
|
|
|
| 18 |
Bu |
3138 |
3013 |
36.73 |
|
|
|
| 19 |
Bu |
3027 |
2907 |
98.92 |
|
|
|
| 20 |
Bu |
1543 |
1482 |
26.21 |
|
|
|
| 21 |
Bu |
1466 |
1408 |
10.56 |
|
|
|
| 22 |
Bu |
1184 |
1137 |
11.09 |
|
|
|
| 23 |
Bu |
1072 |
1030 |
106.72 |
|
|
|
| 24 |
Bu |
304 |
292 |
11.16 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18327.6 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 17600.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G*
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
-0.440 |
0.587 |
0.000 |
| O2 |
0.440 |
-0.587 |
0.000 |
| C3 |
0.440 |
1.691 |
0.000 |
| C4 |
-0.440 |
-1.691 |
0.000 |
| H5 |
-0.215 |
2.567 |
0.000 |
| H6 |
1.073 |
1.701 |
0.897 |
| H7 |
1.073 |
1.701 |
-0.897 |
| H8 |
0.215 |
-2.567 |
0.000 |
| H9 |
-1.073 |
-1.701 |
0.897 |
| H10 |
-1.073 |
-1.701 |
-0.897 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| O1 | | 1.4677 | 1.4121 | 2.2781 | 1.9928 | 2.0818 | 2.0818 | 3.2218 | 2.5374 | 2.5374 |
O2 | 1.4677 | | 2.2781 | 1.4121 | 3.2218 | 2.5374 | 2.5374 | 1.9928 | 2.0818 | 2.0818 | C3 | 1.4121 | 2.2781 | | 3.4948 | 1.0944 | 1.0977 | 1.0977 | 4.2642 | 3.8206 | 3.8206 | C4 | 2.2781 | 1.4121 | 3.4948 | | 4.2642 | 3.8206 | 3.8206 | 1.0944 | 1.0977 | 1.0977 | H5 | 1.9928 | 3.2218 | 1.0944 | 4.2642 | | 1.7932 | 1.7932 | 5.1525 | 4.4445 | 4.4445 | H6 | 2.0818 | 2.5374 | 1.0977 | 3.8206 | 1.7932 | | 1.7944 | 4.4445 | 4.0212 | 4.4035 | H7 | 2.0818 | 2.5374 | 1.0977 | 3.8206 | 1.7932 | 1.7944 | | 4.4445 | 4.4035 | 4.0212 | H8 | 3.2218 | 1.9928 | 4.2642 | 1.0944 | 5.1525 | 4.4445 | 4.4445 | | 1.7932 | 1.7932 | H9 | 2.5374 | 2.0818 | 3.8206 | 1.0977 | 4.4445 | 4.0212 | 4.4035 | 1.7932 | | 1.7944 | H10 | 2.5374 | 2.0818 | 3.8206 | 1.0977 | 4.4445 | 4.4035 | 4.0212 | 1.7932 | 1.7944 | |
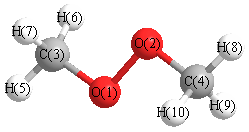 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
O2 |
C4 |
104.554 |
|
O1 |
C3 |
H5 |
104.608 |
| O1 |
C3 |
H6 |
111.472 |
|
O1 |
C3 |
H7 |
111.472 |
| O2 |
O1 |
C3 |
104.554 |
|
O2 |
C4 |
H8 |
104.608 |
| O2 |
C4 |
H9 |
111.472 |
|
O2 |
C4 |
H10 |
111.472 |
| H5 |
C3 |
H6 |
109.769 |
|
H5 |
C3 |
H7 |
109.769 |
| H6 |
C3 |
H7 |
109.638 |
|
H8 |
C4 |
H9 |
109.769 |
| H8 |
C4 |
H10 |
109.769 |
|
H9 |
C4 |
H10 |
109.638 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.282 |
|
|
|
| 2 |
O |
-0.282 |
|
|
|
| 3 |
C |
-0.177 |
|
|
|
| 4 |
C |
-0.177 |
|
|
|
| 5 |
H |
0.161 |
|
|
|
| 6 |
H |
0.149 |
|
|
|
| 7 |
H |
0.149 |
|
|
|
| 8 |
H |
0.161 |
|
|
|
| 9 |
H |
0.149 |
|
|
|
| 10 |
H |
0.149 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.325 |
1.704 |
0.000 |
| y |
1.704 |
-18.520 |
0.000 |
| z |
0.000 |
0.000 |
-25.036 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.547 |
1.704 |
0.000 |
| y |
1.704 |
6.661 |
0.000 |
| z |
0.000 |
0.000 |
-3.114 |
|
| Polar |
|---|
| 3z2-r2 | -6.228 |
|---|
| x2-y2 | -6.805 |
|---|
| xy | 1.704 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.151 |
0.192 |
0.000 |
| y |
0.192 |
6.520 |
0.000 |
| z |
0.000 |
0.000 |
3.879 |
<r2> (average value of r
2) Å
2
| <r2> |
92.263 |
| (<r2>)1/2 |
9.605 |
Jump to
S1C1
Energy calculated at B3LYP/6-31G*
| | hartrees |
|---|
| Energy at 0K | -230.161127 |
| Energy at 298.15K | -230.168189 |
| Nuclear repulsion energy | 134.023569 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3131 |
3007 |
1.48 |
|
|
|
| 2 |
A |
3095 |
2972 |
78.48 |
|
|
|
| 3 |
A |
3030 |
2910 |
18.73 |
|
|
|
| 4 |
A |
1546 |
1484 |
0.00 |
|
|
|
| 5 |
A |
1493 |
1434 |
6.03 |
|
|
|
| 6 |
A |
1480 |
1421 |
0.02 |
|
|
|
| 7 |
A |
1255 |
1205 |
0.55 |
|
|
|
| 8 |
A |
1188 |
1141 |
0.99 |
|
|
|
| 9 |
A |
1066 |
1024 |
7.01 |
|
|
|
| 10 |
A |
842 |
809 |
2.28 |
|
|
|
| 11 |
A |
461 |
442 |
3.11 |
|
|
|
| 12 |
A |
234 |
225 |
1.95 |
|
|
|
| 13 |
A |
31 |
30 |
8.45 |
|
|
|
| 14 |
B |
3131 |
3007 |
39.93 |
|
|
|
| 15 |
B |
3093 |
2970 |
23.59 |
|
|
|
| 16 |
B |
3026 |
2906 |
69.65 |
|
|
|
| 17 |
B |
1539 |
1478 |
22.50 |
|
|
|
| 18 |
B |
1493 |
1433 |
1.59 |
|
|
|
| 19 |
B |
1467 |
1409 |
12.17 |
|
|
|
| 20 |
B |
1201 |
1154 |
3.95 |
|
|
|
| 21 |
B |
1184 |
1137 |
5.65 |
|
|
|
| 22 |
B |
1053 |
1011 |
78.06 |
|
|
|
| 23 |
B |
405 |
389 |
7.39 |
|
|
|
| 24 |
B |
242 |
233 |
4.12 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18343.5 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 17615.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
-0.405 |
0.609 |
-0.344 |
| O2 |
0.405 |
-0.609 |
-0.344 |
| C3 |
0.405 |
1.602 |
0.257 |
| C4 |
-0.405 |
-1.602 |
0.257 |
| H5 |
-0.201 |
2.514 |
0.216 |
| H6 |
0.641 |
1.360 |
1.301 |
| H7 |
1.335 |
1.755 |
-0.305 |
| H8 |
0.201 |
-2.514 |
0.216 |
| H9 |
-0.641 |
-1.360 |
1.301 |
| H10 |
-1.335 |
-1.755 |
-0.305 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| O1 | | 1.4621 | 1.4152 | 2.2912 | 1.9958 | 2.0890 | 2.0833 | 3.2296 | 2.5765 | 2.5406 |
O2 | 1.4621 | | 2.2912 | 1.4152 | 3.2296 | 2.5765 | 2.5406 | 1.9958 | 2.0890 | 2.0833 | C3 | 1.4152 | 2.2912 | | 3.3050 | 1.0951 | 1.0980 | 1.0971 | 4.1210 | 3.3102 | 3.8225 | C4 | 2.2912 | 1.4152 | 3.3050 | | 4.1210 | 3.3102 | 3.8225 | 1.0951 | 1.0980 | 1.0971 | H5 | 1.9958 | 3.2296 | 1.0951 | 4.1210 | | 1.7937 | 1.7904 | 5.0432 | 4.0464 | 4.4472 | H6 | 2.0890 | 2.5765 | 1.0980 | 3.3102 | 1.7937 | | 1.7939 | 4.0464 | 3.0063 | 4.0230 | H7 | 2.0833 | 2.5406 | 1.0971 | 3.8225 | 1.7904 | 1.7939 | | 4.4472 | 4.0230 | 4.4097 | H8 | 3.2296 | 1.9958 | 4.1210 | 1.0951 | 5.0432 | 4.0464 | 4.4472 | | 1.7937 | 1.7904 | H9 | 2.5765 | 2.0890 | 3.3102 | 1.0980 | 4.0464 | 3.0063 | 4.0230 | 1.7937 | | 1.7939 | H10 | 2.5406 | 2.0833 | 3.8225 | 1.0971 | 4.4472 | 4.0230 | 4.4097 | 1.7904 | 1.7939 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
O2 |
C4 |
105.550 |
|
O1 |
C3 |
H5 |
104.598 |
| O1 |
C3 |
H6 |
111.828 |
|
O1 |
C3 |
H7 |
111.416 |
| O2 |
O1 |
C3 |
105.550 |
|
O2 |
C4 |
H8 |
104.598 |
| O2 |
C4 |
H9 |
111.828 |
|
O2 |
C4 |
H10 |
111.416 |
| H5 |
C3 |
H6 |
109.747 |
|
H5 |
C3 |
H7 |
109.515 |
| H6 |
C3 |
H7 |
109.614 |
|
H8 |
C4 |
H9 |
109.747 |
| H8 |
C4 |
H10 |
109.515 |
|
H9 |
C4 |
H10 |
109.614 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.278 |
|
|
|
| 2 |
O |
-0.278 |
|
|
|
| 3 |
C |
-0.176 |
|
|
|
| 4 |
C |
-0.176 |
|
|
|
| 5 |
H |
0.159 |
|
|
|
| 6 |
H |
0.142 |
|
|
|
| 7 |
H |
0.153 |
|
|
|
| 8 |
H |
0.159 |
|
|
|
| 9 |
H |
0.142 |
|
|
|
| 10 |
H |
0.153 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.339 |
1.339 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.235 |
1.638 |
0.000 |
| y |
1.638 |
-19.253 |
0.000 |
| z |
0.000 |
0.000 |
-25.371 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.923 |
1.638 |
0.000 |
| y |
1.638 |
6.050 |
0.000 |
| z |
0.000 |
0.000 |
-3.127 |
|
| Polar |
|---|
| 3z2-r2 | -6.254 |
|---|
| x2-y2 | -5.982 |
|---|
| xy | 1.638 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.087 |
0.137 |
0.000 |
| y |
0.137 |
6.388 |
0.000 |
| z |
0.000 |
0.000 |
3.890 |
<r2> (average value of r
2) Å
2
| <r2> |
89.174 |
| (<r2>)1/2 |
9.443 |