Jump to
S1C2
Energy calculated at B3LYP/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -35.989550 |
| Energy at 298.15K | |
| HF Energy | -35.989550 |
| Nuclear repulsion energy | 29.786048 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3273 |
3169 |
2.54 |
|
|
|
| 2 |
A1 |
682 |
660 |
16.26 |
|
|
|
| 3 |
A1 |
284 |
275 |
0.47 |
|
|
|
| 4 |
B1 |
407i |
395i |
128.29 |
|
|
|
| 5 |
B2 |
1195 |
1157 |
56.42 |
|
|
|
| 6 |
B2 |
854 |
827 |
173.51 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 2939.8 cm
-1
Scaled (by 0.9684) Zero Point Vibrational Energy (zpe) 2846.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
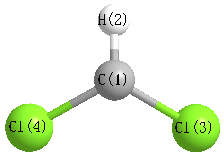
Geometric Data calculated at B3LYP/CEP-31G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.712 |
| H2 |
0.000 |
0.000 |
1.801 |
| Cl3 |
0.000 |
1.553 |
-0.179 |
| Cl4 |
0.000 |
-1.553 |
-0.179 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
Cl3 |
Cl4 |
| C1 | | 1.0895 | 1.7903 | 1.7903 |
H2 | 1.0895 | | 2.5163 | 2.5163 | Cl3 | 1.7903 | 2.5163 | | 3.1067 | Cl4 | 1.7903 | 2.5163 | 3.1067 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Cl3 |
C1 |
H2 |
119.815 |
|
Cl3 |
C1 |
Cl4 |
120.370 |
| Cl4 |
C1 |
H2 |
119.815 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.376 |
|
|
|
| 2 |
H |
0.278 |
|
|
|
| 3 |
Cl |
0.049 |
|
|
|
| 4 |
Cl |
0.049 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.325 |
1.325 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.085 |
0.000 |
0.000 |
| y |
0.000 |
-31.289 |
0.000 |
| z |
0.000 |
0.000 |
-27.520 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.681 |
0.000 |
0.000 |
| y |
0.000 |
-1.986 |
0.000 |
| z |
0.000 |
0.000 |
3.667 |
|
| Polar |
|---|
| 3z2-r2 | 7.334 |
|---|
| x2-y2 | 0.204 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.180 |
0.000 |
0.000 |
| y |
0.000 |
6.944 |
0.000 |
| z |
0.000 |
0.000 |
2.935 |
<r2> (average value of r
2) Å
2
| <r2> |
58.211 |
| (<r2>)1/2 |
7.630 |
Jump to
S1C1
Energy calculated at B3LYP/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -35.990943 |
| Energy at 298.15K | -35.991670 |
| HF Energy | -35.990943 |
| Nuclear repulsion energy | 29.683054 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3223 |
3121 |
9.37 |
|
|
|
| 2 |
A' |
701 |
679 |
31.46 |
|
|
|
| 3 |
A' |
517 |
501 |
32.98 |
|
|
|
| 4 |
A' |
278 |
269 |
0.55 |
|
|
|
| 5 |
A" |
1208 |
1170 |
44.82 |
|
|
|
| 6 |
A" |
810 |
784 |
207.57 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3368.6 cm
-1
Scaled (by 0.9684) Zero Point Vibrational Energy (zpe) 3262.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/CEP-31G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.014 |
0.744 |
0.000 |
| H2 |
-0.562 |
1.674 |
0.000 |
| Cl3 |
0.014 |
-0.181 |
1.551 |
| Cl4 |
0.014 |
-0.181 |
-1.551 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
Cl3 |
Cl4 |
| C1 | | 1.0941 | 1.8059 | 1.8059 |
H2 | 1.0941 | | 2.4858 | 2.4858 | Cl3 | 1.8059 | 2.4858 | | 3.1020 | Cl4 | 1.8059 | 2.4858 | 3.1020 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Cl3 |
C1 |
H2 |
115.809 |
|
Cl3 |
C1 |
Cl4 |
118.372 |
| Cl4 |
C1 |
H2 |
115.809 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.320 |
|
|
|
| 2 |
H |
0.253 |
|
|
|
| 3 |
Cl |
0.033 |
|
|
|
| 4 |
Cl |
0.033 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.859 |
1.213 |
0.000 |
1.486 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.651 |
-1.309 |
0.000 |
| y |
-1.309 |
-28.204 |
0.000 |
| z |
0.000 |
0.000 |
-31.646 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.726 |
-1.309 |
0.000 |
| y |
-1.309 |
2.945 |
0.000 |
| z |
0.000 |
0.000 |
-2.219 |
|
| Polar |
|---|
| 3z2-r2 | -4.438 |
|---|
| x2-y2 | -2.447 |
|---|
| xy | -1.309 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.228 |
-0.142 |
0.000 |
| y |
-0.142 |
3.073 |
0.000 |
| z |
0.000 |
0.000 |
7.103 |
<r2> (average value of r
2) Å
2
| <r2> |
58.318 |
| (<r2>)1/2 |
7.637 |