Jump to
S1C2
Energy calculated at B3LYP/TZVP
| | hartrees |
|---|
| Energy at 0K | -999.099380 |
| Energy at 298.15K | -999.104028 |
| HF Energy | -999.099380 |
| Nuclear repulsion energy | 192.707657 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3105 |
2998 |
0.00 |
|
|
|
| 2 |
Ag |
1497 |
1445 |
0.00 |
|
|
|
| 3 |
Ag |
1343 |
1297 |
0.00 |
|
|
|
| 4 |
Ag |
1064 |
1027 |
0.00 |
|
|
|
| 5 |
Ag |
741 |
716 |
0.00 |
|
|
|
| 6 |
Ag |
294 |
284 |
0.00 |
|
|
|
| 7 |
Au |
3184 |
3074 |
3.62 |
|
|
|
| 8 |
Au |
1143 |
1104 |
1.86 |
|
|
|
| 9 |
Au |
787 |
760 |
2.21 |
|
|
|
| 10 |
Au |
119 |
114 |
7.38 |
|
|
|
| 11 |
Bg |
3161 |
3052 |
0.00 |
|
|
|
| 12 |
Bg |
1300 |
1255 |
0.00 |
|
|
|
| 13 |
Bg |
1009 |
974 |
0.00 |
|
|
|
| 14 |
Bu |
3114 |
3006 |
14.70 |
|
|
|
| 15 |
Bu |
1498 |
1446 |
6.52 |
|
|
|
| 16 |
Bu |
1269 |
1225 |
41.64 |
|
|
|
| 17 |
Bu |
696 |
672 |
108.89 |
|
|
|
| 18 |
Bu |
213 |
206 |
10.87 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 12768.9 cm
-1
Scaled (by 0.9654) Zero Point Vibrational Energy (zpe) 12327.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/TZVP
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.478 |
0.585 |
0.000 |
| C2 |
-0.478 |
-0.585 |
0.000 |
| Cl3 |
-0.478 |
2.137 |
0.000 |
| Cl4 |
0.478 |
-2.137 |
0.000 |
| H5 |
1.101 |
0.597 |
0.890 |
| H6 |
1.101 |
0.597 |
-0.890 |
| H7 |
-1.101 |
-0.597 |
0.890 |
| H8 |
-1.101 |
-0.597 |
-0.890 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
Cl3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.5108 | 1.8230 | 2.7214 | 1.0860 | 1.0860 | 2.1639 | 2.1639 |
C2 | 1.5108 | | 2.7214 | 1.8230 | 2.1639 | 2.1639 | 1.0860 | 1.0860 | Cl3 | 1.8230 | 2.7214 | | 4.3790 | 2.3782 | 2.3782 | 2.9415 | 2.9415 | Cl4 | 2.7214 | 1.8230 | 4.3790 | | 2.9415 | 2.9415 | 2.3782 | 2.3782 | H5 | 1.0860 | 2.1639 | 2.3782 | 2.9415 | | 1.7790 | 2.5053 | 3.0727 | H6 | 1.0860 | 2.1639 | 2.3782 | 2.9415 | 1.7790 | | 3.0727 | 2.5053 | H7 | 2.1639 | 1.0860 | 2.9415 | 2.3782 | 2.5053 | 3.0727 | | 1.7790 | H8 | 2.1639 | 1.0860 | 2.9415 | 2.3782 | 3.0727 | 2.5053 | 1.7790 | |
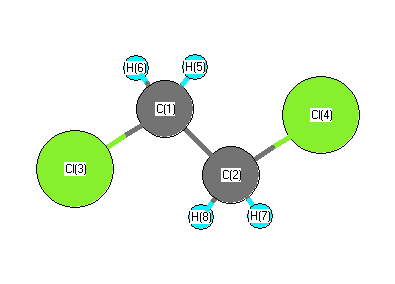 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
Cl4 |
109.072 |
|
C1 |
C2 |
H7 |
111.838 |
| C1 |
C2 |
H8 |
111.838 |
|
C2 |
C1 |
Cl3 |
109.072 |
| C2 |
C1 |
H5 |
111.838 |
|
C2 |
C1 |
H6 |
111.838 |
| Cl3 |
C1 |
H5 |
106.931 |
|
Cl3 |
C1 |
H6 |
106.931 |
| Cl4 |
C2 |
H7 |
106.931 |
|
Cl4 |
C2 |
H8 |
106.931 |
| H5 |
C1 |
H6 |
109.984 |
|
H7 |
C2 |
H8 |
109.984 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.200 |
|
|
|
| 2 |
C |
-0.200 |
|
|
|
| 3 |
Cl |
-0.132 |
|
|
|
| 4 |
Cl |
-0.132 |
|
|
|
| 5 |
H |
0.166 |
|
|
|
| 6 |
H |
0.166 |
|
|
|
| 7 |
H |
0.166 |
|
|
|
| 8 |
H |
0.166 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.542 |
2.121 |
0.000 |
| y |
2.121 |
-45.747 |
0.000 |
| z |
0.000 |
0.000 |
-37.816 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.240 |
2.121 |
0.000 |
| y |
2.121 |
-8.068 |
0.000 |
| z |
0.000 |
0.000 |
3.829 |
|
| Polar |
|---|
| 3z2-r2 | 7.658 |
|---|
| x2-y2 | 8.205 |
|---|
| xy | 2.121 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.928 |
-1.500 |
0.000 |
| y |
-1.500 |
9.596 |
0.000 |
| z |
0.000 |
0.000 |
5.012 |
<r2> (average value of r
2) Å
2
| <r2> |
204.497 |
| (<r2>)1/2 |
14.300 |
Jump to
S1C1
Energy calculated at B3LYP/TZVP
| | hartrees |
|---|
| Energy at 0K | -999.096573 |
| Energy at 298.15K | -999.101359 |
| HF Energy | -999.096573 |
| Nuclear repulsion energy | 199.924831 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3143 |
3034 |
0.53 |
|
|
|
| 2 |
A |
3088 |
2981 |
22.45 |
|
|
|
| 3 |
A |
1475 |
1424 |
0.98 |
|
|
|
| 4 |
A |
1349 |
1302 |
21.52 |
|
|
|
| 5 |
A |
1229 |
1187 |
1.11 |
|
|
|
| 6 |
A |
1048 |
1012 |
1.17 |
|
|
|
| 7 |
A |
951 |
918 |
11.40 |
|
|
|
| 8 |
A |
645 |
623 |
22.58 |
|
|
|
| 9 |
A |
260 |
251 |
0.91 |
|
|
|
| 10 |
A |
112 |
108 |
1.02 |
|
|
|
| 11 |
B |
3157 |
3048 |
5.00 |
|
|
|
| 12 |
B |
3080 |
2973 |
2.91 |
|
|
|
| 13 |
B |
1471 |
1420 |
12.15 |
|
|
|
| 14 |
B |
1326 |
1280 |
46.92 |
|
|
|
| 15 |
B |
1163 |
1122 |
1.11 |
|
|
|
| 16 |
B |
895 |
865 |
18.97 |
|
|
|
| 17 |
B |
669 |
645 |
30.76 |
|
|
|
| 18 |
B |
407 |
393 |
9.78 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 12733.1 cm
-1
Scaled (by 0.9654) Zero Point Vibrational Energy (zpe) 12292.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/TZVP
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.299 |
0.692 |
0.898 |
| C2 |
-0.299 |
-0.692 |
0.898 |
| Cl3 |
-0.299 |
1.722 |
-0.472 |
| Cl4 |
0.299 |
-1.722 |
-0.472 |
| H5 |
0.013 |
1.207 |
1.814 |
| H6 |
1.383 |
0.662 |
0.820 |
| H7 |
-0.013 |
-1.207 |
1.814 |
| H8 |
-1.383 |
-0.662 |
0.820 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
Cl3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.5080 | 1.8150 | 2.7758 | 1.0894 | 1.0867 | 2.1316 | 2.1605 |
C2 | 1.5080 | | 2.7758 | 1.8150 | 2.1316 | 2.1605 | 1.0894 | 1.0867 | Cl3 | 1.8150 | 2.7758 | | 3.4964 | 2.3640 | 2.3711 | 3.7266 | 2.9206 | Cl4 | 2.7758 | 1.8150 | 3.4964 | | 3.7266 | 2.9206 | 2.3640 | 2.3711 | H5 | 1.0894 | 2.1316 | 2.3640 | 3.7266 | | 1.7776 | 2.4138 | 2.5353 | H6 | 1.0867 | 2.1605 | 2.3711 | 2.9206 | 1.7776 | | 2.5353 | 3.0660 | H7 | 2.1316 | 1.0894 | 3.7266 | 2.3640 | 2.4138 | 2.5353 | | 1.7776 | H8 | 2.1605 | 1.0867 | 2.9206 | 2.3711 | 2.5353 | 3.0660 | 1.7776 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
Cl4 |
112.979 |
|
C1 |
C2 |
H7 |
109.241 |
| C1 |
C2 |
H8 |
111.719 |
|
C2 |
C1 |
Cl3 |
112.979 |
| C2 |
C1 |
H5 |
109.241 |
|
C2 |
C1 |
H6 |
111.719 |
| Cl3 |
C1 |
H5 |
106.265 |
|
Cl3 |
C1 |
H6 |
106.906 |
| Cl4 |
C2 |
H7 |
106.265 |
|
Cl4 |
C2 |
H8 |
106.906 |
| H5 |
C1 |
H6 |
109.543 |
|
H7 |
C2 |
H8 |
109.543 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.200 |
|
|
|
| 2 |
C |
-0.200 |
|
|
|
| 3 |
Cl |
-0.121 |
|
|
|
| 4 |
Cl |
-0.121 |
|
|
|
| 5 |
H |
0.156 |
|
|
|
| 6 |
H |
0.166 |
|
|
|
| 7 |
H |
0.156 |
|
|
|
| 8 |
H |
0.166 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
2.909 |
2.909 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.709 |
1.266 |
0.000 |
| y |
1.266 |
-42.890 |
0.000 |
| z |
0.000 |
0.000 |
-35.510 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.491 |
1.266 |
0.000 |
| y |
1.266 |
-6.281 |
0.000 |
| z |
0.000 |
0.000 |
4.790 |
|
| Polar |
|---|
| 3z2-r2 | 9.579 |
|---|
| x2-y2 | 5.181 |
|---|
| xy | 1.266 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.374 |
-0.585 |
0.000 |
| y |
-0.585 |
7.538 |
0.000 |
| z |
0.000 |
0.000 |
7.039 |
<r2> (average value of r
2) Å
2
| <r2> |
167.680 |
| (<r2>)1/2 |
12.949 |