Jump to
S1C2
Energy calculated at B3LYP/TZVP
| | hartrees |
|---|
| Energy at 0K | -345.441333 |
| Energy at 298.15K | -345.458435 |
| HF Energy | -345.441333 |
| Nuclear repulsion energy | 424.379876 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1' |
3042 |
2937 |
0.00 |
|
|
|
| 2 |
A1' |
1504 |
1452 |
0.00 |
|
|
|
| 3 |
A1' |
1358 |
1311 |
0.00 |
|
|
|
| 4 |
A1' |
954 |
921 |
0.00 |
|
|
|
| 5 |
A1' |
808 |
780 |
0.00 |
|
|
|
| 6 |
A1' |
610 |
589 |
0.00 |
|
|
|
| 7 |
A1" |
3058 |
2953 |
0.00 |
|
|
|
| 8 |
A1" |
1265 |
1221 |
0.00 |
|
|
|
| 9 |
A1" |
1025 |
989 |
0.00 |
|
|
|
| 10 |
A1" |
70 |
68 |
0.00 |
|
|
|
| 11 |
A2' |
3080 |
2974 |
0.00 |
|
|
|
| 12 |
A2' |
1198 |
1157 |
0.00 |
|
|
|
| 13 |
A2' |
813 |
785 |
0.00 |
|
|
|
| 14 |
A2" |
3029 |
2924 |
113.82 |
|
|
|
| 15 |
A2" |
1496 |
1444 |
7.63 |
|
|
|
| 16 |
A2" |
1386 |
1338 |
3.75 |
|
|
|
| 17 |
A2" |
995 |
960 |
17.69 |
|
|
|
| 18 |
A2" |
757 |
731 |
65.38 |
|
|
|
| 19 |
E' |
3088 |
2981 |
100.48 |
|
|
|
| 19 |
E' |
3088 |
2981 |
100.46 |
|
|
|
| 20 |
E' |
3034 |
2929 |
119.38 |
|
|
|
| 20 |
E' |
3034 |
2929 |
119.40 |
|
|
|
| 21 |
E' |
1497 |
1445 |
11.38 |
|
|
|
| 21 |
E' |
1497 |
1445 |
11.38 |
|
|
|
| 22 |
E' |
1353 |
1307 |
9.16 |
|
|
|
| 22 |
E' |
1353 |
1307 |
9.15 |
|
|
|
| 23 |
E' |
1329 |
1283 |
0.07 |
|
|
|
| 23 |
E' |
1329 |
1283 |
0.07 |
|
|
|
| 24 |
E' |
1068 |
1031 |
43.07 |
|
|
|
| 24 |
E' |
1068 |
1031 |
43.07 |
|
|
|
| 25 |
E' |
888 |
857 |
3.40 |
|
|
|
| 25 |
E' |
888 |
857 |
3.40 |
|
|
|
| 26 |
E' |
829 |
800 |
4.88 |
|
|
|
| 26 |
E' |
829 |
800 |
4.88 |
|
|
|
| 27 |
E' |
425 |
411 |
0.00 |
|
|
|
| 27 |
E' |
425 |
411 |
0.00 |
|
|
|
| 28 |
E" |
3062 |
2956 |
0.00 |
|
|
|
| 28 |
E" |
3062 |
2956 |
0.00 |
|
|
|
| 29 |
E" |
3026 |
2921 |
0.00 |
|
|
|
| 29 |
E" |
3026 |
2921 |
0.00 |
|
|
|
| 30 |
E" |
1486 |
1434 |
0.00 |
|
|
|
| 30 |
E" |
1486 |
1434 |
0.00 |
|
|
|
| 31 |
E" |
1343 |
1297 |
0.00 |
|
|
|
| 31 |
E" |
1343 |
1297 |
0.00 |
|
|
|
| 32 |
E" |
1328 |
1282 |
0.00 |
|
|
|
| 32 |
E" |
1328 |
1282 |
0.00 |
|
|
|
| 33 |
E" |
1206 |
1164 |
0.00 |
|
|
|
| 33 |
E" |
1206 |
1164 |
0.00 |
|
|
|
| 34 |
E" |
1032 |
996 |
0.00 |
|
|
|
| 34 |
E" |
1032 |
996 |
0.00 |
|
|
|
| 35 |
E" |
587 |
567 |
0.00 |
|
|
|
| 35 |
E" |
587 |
567 |
0.00 |
|
|
|
| 36 |
E" |
331 |
320 |
0.00 |
|
|
|
| 36 |
E" |
331 |
320 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40134.6 cm
-1
Scaled (by 0.9654) Zero Point Vibrational Energy (zpe) 38746.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/TZVP
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.283 |
| N2 |
0.000 |
0.000 |
-1.283 |
| C3 |
0.000 |
1.383 |
0.781 |
| C4 |
1.198 |
-0.692 |
0.781 |
| C5 |
-1.198 |
-0.692 |
0.781 |
| C6 |
0.000 |
1.383 |
-0.781 |
| C7 |
-1.198 |
-0.692 |
-0.781 |
| C8 |
1.198 |
-0.692 |
-0.781 |
| H9 |
0.881 |
1.891 |
1.182 |
| H10 |
-0.881 |
1.891 |
1.182 |
| H11 |
1.198 |
-1.708 |
1.182 |
| H12 |
2.078 |
-0.183 |
1.182 |
| H13 |
-2.078 |
-0.183 |
1.182 |
| H14 |
-1.198 |
-1.708 |
1.182 |
| H15 |
-0.881 |
1.891 |
-1.182 |
| H16 |
0.881 |
1.891 |
-1.182 |
| H17 |
-1.198 |
-1.708 |
-1.182 |
| H18 |
-2.078 |
-0.183 |
-1.182 |
| H19 |
2.078 |
-0.183 |
-1.182 |
| H20 |
1.198 |
-1.708 |
-1.182 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5664 | 1.4714 | 1.4714 | 1.4714 | 2.4851 | 2.4851 | 2.4851 | 2.0888 | 2.0888 | 2.0888 | 2.0888 | 2.0888 | 2.0888 | 3.2295 | 3.2295 | 3.2295 | 3.2295 | 3.2295 | 3.2295 |
N2 | 2.5664 | | 2.4851 | 2.4851 | 2.4851 | 1.4714 | 1.4714 | 1.4714 | 3.2295 | 3.2295 | 3.2295 | 3.2295 | 3.2295 | 3.2295 | 2.0888 | 2.0888 | 2.0888 | 2.0888 | 2.0888 | 2.0888 | C3 | 1.4714 | 2.4851 | | 2.3958 | 2.3958 | 1.5628 | 2.8604 | 2.8604 | 1.0927 | 1.0927 | 3.3395 | 2.6331 | 2.6331 | 3.3395 | 2.2109 | 2.2109 | 3.8531 | 3.2600 | 3.2600 | 3.8531 | C4 | 1.4714 | 2.4851 | 2.3958 | | 2.3958 | 2.8604 | 2.8604 | 1.5628 | 2.6331 | 3.3395 | 1.0927 | 1.0927 | 3.3395 | 2.6331 | 3.8531 | 3.2600 | 3.2600 | 3.8531 | 2.2109 | 2.2109 | C5 | 1.4714 | 2.4851 | 2.3958 | 2.3958 | | 2.8604 | 1.5628 | 2.8604 | 3.3395 | 2.6331 | 2.6331 | 3.3395 | 1.0927 | 1.0927 | 3.2600 | 3.8531 | 2.2109 | 2.2109 | 3.8531 | 3.2600 | C6 | 2.4851 | 1.4714 | 1.5628 | 2.8604 | 2.8604 | | 2.3958 | 2.3958 | 2.2109 | 2.2109 | 3.8531 | 3.2600 | 3.2600 | 3.8531 | 1.0927 | 1.0927 | 3.3395 | 2.6331 | 2.6331 | 3.3395 | C7 | 2.4851 | 1.4714 | 2.8604 | 2.8604 | 1.5628 | 2.3958 | | 2.3958 | 3.8531 | 3.2600 | 3.2600 | 3.8531 | 2.2109 | 2.2109 | 2.6331 | 3.3395 | 1.0927 | 1.0927 | 3.3395 | 2.6331 | C8 | 2.4851 | 1.4714 | 2.8604 | 1.5628 | 2.8604 | 2.3958 | 2.3958 | | 3.2600 | 3.8531 | 2.2109 | 2.2109 | 3.8531 | 3.2600 | 3.3395 | 2.6331 | 2.6331 | 3.3395 | 1.0927 | 1.0927 | H9 | 2.0888 | 3.2295 | 1.0927 | 2.6331 | 3.3395 | 2.2109 | 3.8531 | 3.2600 | | 1.7611 | 3.6137 | 2.3956 | 3.6137 | 4.1566 | 2.9477 | 2.3638 | 4.7818 | 4.3181 | 3.3655 | 4.3181 | H10 | 2.0888 | 3.2295 | 1.0927 | 3.3395 | 2.6331 | 2.2109 | 3.2600 | 3.8531 | 1.7611 | | 4.1566 | 3.6137 | 2.3956 | 3.6137 | 2.3638 | 2.9477 | 4.3181 | 3.3655 | 4.3181 | 4.7818 | H11 | 2.0888 | 3.2295 | 3.3395 | 1.0927 | 2.6331 | 3.8531 | 3.2600 | 2.2109 | 3.6137 | 4.1566 | | 1.7611 | 3.6137 | 2.3956 | 4.7818 | 4.3181 | 3.3655 | 4.3181 | 2.9477 | 2.3638 | H12 | 2.0888 | 3.2295 | 2.6331 | 1.0927 | 3.3395 | 3.2600 | 3.8531 | 2.2109 | 2.3956 | 3.6137 | 1.7611 | | 4.1566 | 3.6137 | 4.3181 | 3.3655 | 4.3181 | 4.7818 | 2.3638 | 2.9477 | H13 | 2.0888 | 3.2295 | 2.6331 | 3.3395 | 1.0927 | 3.2600 | 2.2109 | 3.8531 | 3.6137 | 2.3956 | 3.6137 | 4.1566 | | 1.7611 | 3.3655 | 4.3181 | 2.9477 | 2.3638 | 4.7818 | 4.3181 | H14 | 2.0888 | 3.2295 | 3.3395 | 2.6331 | 1.0927 | 3.8531 | 2.2109 | 3.2600 | 4.1566 | 3.6137 | 2.3956 | 3.6137 | 1.7611 | | 4.3181 | 4.7818 | 2.3638 | 2.9477 | 4.3181 | 3.3655 | H15 | 3.2295 | 2.0888 | 2.2109 | 3.8531 | 3.2600 | 1.0927 | 2.6331 | 3.3395 | 2.9477 | 2.3638 | 4.7818 | 4.3181 | 3.3655 | 4.3181 | | 1.7611 | 3.6137 | 2.3956 | 3.6137 | 4.1566 | H16 | 3.2295 | 2.0888 | 2.2109 | 3.2600 | 3.8531 | 1.0927 | 3.3395 | 2.6331 | 2.3638 | 2.9477 | 4.3181 | 3.3655 | 4.3181 | 4.7818 | 1.7611 | | 4.1566 | 3.6137 | 2.3956 | 3.6137 | H17 | 3.2295 | 2.0888 | 3.8531 | 3.2600 | 2.2109 | 3.3395 | 1.0927 | 2.6331 | 4.7818 | 4.3181 | 3.3655 | 4.3181 | 2.9477 | 2.3638 | 3.6137 | 4.1566 | | 1.7611 | 3.6137 | 2.3956 | H18 | 3.2295 | 2.0888 | 3.2600 | 3.8531 | 2.2109 | 2.6331 | 1.0927 | 3.3395 | 4.3181 | 3.3655 | 4.3181 | 4.7818 | 2.3638 | 2.9477 | 2.3956 | 3.6137 | 1.7611 | | 4.1566 | 3.6137 | H19 | 3.2295 | 2.0888 | 3.2600 | 2.2109 | 3.8531 | 2.6331 | 3.3395 | 1.0927 | 3.3655 | 4.3181 | 2.9477 | 2.3638 | 4.7818 | 4.3181 | 3.6137 | 2.3956 | 3.6137 | 4.1566 | | 1.7611 | H20 | 3.2295 | 2.0888 | 3.8531 | 2.2109 | 3.2600 | 3.3395 | 2.6331 | 1.0927 | 4.3181 | 4.7818 | 2.3638 | 2.9477 | 4.3181 | 3.3655 | 4.1566 | 3.6137 | 2.3956 | 3.6137 | 1.7611 | |
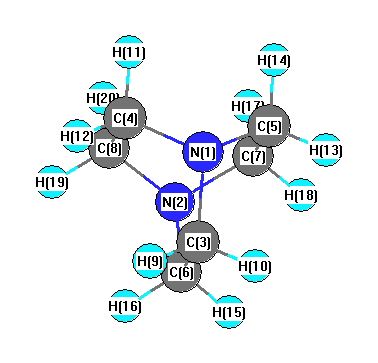 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.940 |
|
N1 |
C3 |
H9 |
108.195 |
| N1 |
C3 |
H10 |
108.195 |
|
N1 |
C4 |
C8 |
109.940 |
| N1 |
C4 |
H11 |
108.195 |
|
N1 |
C4 |
H12 |
108.195 |
| N1 |
C5 |
C7 |
109.940 |
|
N1 |
C5 |
H13 |
108.195 |
| N1 |
C5 |
H14 |
108.195 |
|
N2 |
C6 |
C3 |
109.940 |
| N2 |
C6 |
H15 |
108.195 |
|
N2 |
C6 |
H16 |
108.195 |
| N2 |
C7 |
C5 |
109.940 |
|
N2 |
C7 |
H17 |
108.195 |
| N2 |
C7 |
H18 |
108.195 |
|
N2 |
C8 |
C4 |
109.940 |
| N2 |
C8 |
H19 |
108.195 |
|
N2 |
C8 |
H20 |
108.195 |
| C3 |
N1 |
C4 |
108.999 |
|
C3 |
N1 |
C5 |
108.999 |
| C3 |
C6 |
H15 |
111.501 |
|
C3 |
C6 |
H16 |
111.501 |
| C4 |
N1 |
C5 |
108.999 |
|
C4 |
C8 |
H19 |
111.501 |
| C4 |
C8 |
H20 |
111.501 |
|
C5 |
C6 |
H15 |
101.547 |
| C5 |
C6 |
H16 |
151.054 |
|
C6 |
N2 |
C7 |
108.999 |
| C6 |
N2 |
C8 |
108.999 |
|
C6 |
C3 |
H9 |
111.501 |
| C6 |
C3 |
H10 |
111.501 |
|
C7 |
N2 |
C8 |
108.999 |
| C7 |
C5 |
H13 |
111.501 |
|
C7 |
C5 |
H14 |
111.501 |
| C8 |
C4 |
H11 |
111.501 |
|
C8 |
C4 |
H12 |
111.501 |
| H9 |
C3 |
H10 |
107.377 |
|
H11 |
C4 |
H12 |
107.377 |
| H13 |
C5 |
H14 |
107.377 |
|
H15 |
C6 |
H16 |
107.377 |
| H17 |
C7 |
H18 |
107.377 |
|
H19 |
C8 |
H20 |
107.377 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.117 |
|
|
|
| 2 |
N |
-0.117 |
|
|
|
| 3 |
C |
-0.224 |
|
|
|
| 4 |
C |
-0.224 |
|
|
|
| 5 |
C |
-0.224 |
|
|
|
| 6 |
C |
-0.224 |
|
|
|
| 7 |
C |
-0.224 |
|
|
|
| 8 |
C |
-0.224 |
|
|
|
| 9 |
H |
0.131 |
|
|
|
| 10 |
H |
0.131 |
|
|
|
| 11 |
H |
0.131 |
|
|
|
| 12 |
H |
0.131 |
|
|
|
| 13 |
H |
0.131 |
|
|
|
| 14 |
H |
0.131 |
|
|
|
| 15 |
H |
0.131 |
|
|
|
| 16 |
H |
0.131 |
|
|
|
| 17 |
H |
0.131 |
|
|
|
| 18 |
H |
0.131 |
|
|
|
| 19 |
H |
0.131 |
|
|
|
| 20 |
H |
0.131 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.397 |
0.000 |
0.000 |
| y |
0.000 |
-47.397 |
0.000 |
| z |
0.000 |
0.000 |
-58.908 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.756 |
0.000 |
0.000 |
| y |
0.000 |
5.756 |
0.000 |
| z |
0.000 |
0.000 |
-11.511 |
|
| Polar |
|---|
| 3z2-r2 | -23.022 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.121 |
0.000 |
0.000 |
| y |
0.000 |
12.122 |
0.000 |
| z |
0.000 |
0.000 |
11.130 |
<r2> (average value of r
2) Å
2
| <r2> |
214.908 |
| (<r2>)1/2 |
14.660 |
Jump to
S1C1
Energy calculated at B3LYP/TZVP
| | hartrees |
|---|
| Energy at 0K | -345.441323 |
| Energy at 298.15K | -345.458397 |
| HF Energy | -345.441323 |
| Nuclear repulsion energy | 424.380257 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3059 |
2953 |
0.00 |
|
|
|
| 2 |
A1 |
3042 |
2937 |
0.00 |
|
|
|
| 3 |
A1 |
1505 |
1453 |
0.00 |
|
|
|
| 4 |
A1 |
1358 |
1311 |
0.00 |
|
|
|
| 5 |
A1 |
1265 |
1221 |
0.00 |
|
|
|
| 6 |
A1 |
1024 |
989 |
0.00 |
|
|
|
| 7 |
A1 |
954 |
921 |
0.00 |
|
|
|
| 8 |
A1 |
808 |
780 |
0.00 |
|
|
|
| 9 |
A1 |
610 |
589 |
0.00 |
|
|
|
| 10 |
A1 |
58 |
56 |
0.00 |
|
|
|
| 11 |
A2 |
3081 |
2974 |
0.01 |
|
|
|
| 12 |
A2 |
3029 |
2924 |
113.86 |
|
|
|
| 13 |
A2 |
1497 |
1445 |
7.61 |
|
|
|
| 14 |
A2 |
1386 |
1338 |
3.78 |
|
|
|
| 15 |
A2 |
1197 |
1156 |
0.00 |
|
|
|
| 16 |
A2 |
995 |
960 |
17.79 |
|
|
|
| 17 |
A2 |
811 |
783 |
0.02 |
|
|
|
| 18 |
A2 |
756 |
730 |
65.24 |
|
|
|
| 19 |
E |
3088 |
2981 |
100.21 |
|
|
|
| 19 |
E |
3088 |
2981 |
100.23 |
|
|
|
| 20 |
E |
3062 |
2956 |
0.03 |
|
|
|
| 20 |
E |
3062 |
2956 |
0.03 |
|
|
|
| 21 |
E |
3034 |
2929 |
119.60 |
|
|
|
| 21 |
E |
3034 |
2929 |
119.59 |
|
|
|
| 22 |
E |
3026 |
2921 |
0.01 |
|
|
|
| 22 |
E |
3026 |
2921 |
0.01 |
|
|
|
| 23 |
E |
1497 |
1445 |
11.39 |
|
|
|
| 23 |
E |
1497 |
1445 |
11.40 |
|
|
|
| 24 |
E |
1485 |
1434 |
0.00 |
|
|
|
| 24 |
E |
1485 |
1434 |
0.00 |
|
|
|
| 25 |
E |
1353 |
1306 |
9.12 |
|
|
|
| 25 |
E |
1353 |
1306 |
9.12 |
|
|
|
| 26 |
E |
1343 |
1296 |
0.00 |
|
|
|
| 26 |
E |
1343 |
1296 |
0.00 |
|
|
|
| 27 |
E |
1329 |
1283 |
0.05 |
|
|
|
| 27 |
E |
1329 |
1283 |
0.05 |
|
|
|
| 28 |
E |
1328 |
1282 |
0.01 |
|
|
|
| 28 |
E |
1328 |
1282 |
0.01 |
|
|
|
| 29 |
E |
1206 |
1164 |
0.00 |
|
|
|
| 29 |
E |
1206 |
1164 |
0.00 |
|
|
|
| 30 |
E |
1068 |
1031 |
43.06 |
|
|
|
| 30 |
E |
1068 |
1031 |
43.06 |
|
|
|
| 31 |
E |
1032 |
996 |
0.01 |
|
|
|
| 31 |
E |
1032 |
996 |
0.01 |
|
|
|
| 32 |
E |
888 |
857 |
3.36 |
|
|
|
| 32 |
E |
888 |
857 |
3.36 |
|
|
|
| 33 |
E |
830 |
801 |
4.95 |
|
|
|
| 33 |
E |
830 |
801 |
4.95 |
|
|
|
| 34 |
E |
587 |
566 |
0.00 |
|
|
|
| 34 |
E |
587 |
566 |
0.00 |
|
|
|
| 35 |
E |
425 |
410 |
0.00 |
|
|
|
| 35 |
E |
425 |
410 |
0.00 |
|
|
|
| 36 |
E |
330 |
318 |
0.00 |
|
|
|
| 36 |
E |
330 |
318 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40124.6 cm
-1
Scaled (by 0.9654) Zero Point Vibrational Energy (zpe) 38736.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/TZVP
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.283 |
| N2 |
0.000 |
0.000 |
-1.283 |
| C3 |
-0.003 |
1.383 |
0.781 |
| C4 |
1.200 |
-0.689 |
0.781 |
| C5 |
-1.196 |
-0.695 |
0.781 |
| C6 |
0.003 |
1.383 |
-0.781 |
| C7 |
-1.200 |
-0.689 |
-0.781 |
| C8 |
1.196 |
-0.695 |
-0.781 |
| H9 |
0.873 |
1.896 |
1.186 |
| H10 |
-0.888 |
1.887 |
1.178 |
| H11 |
1.206 |
-1.704 |
1.186 |
| H12 |
2.079 |
-0.174 |
1.178 |
| H13 |
-2.078 |
-0.192 |
1.186 |
| H14 |
-1.190 |
-1.713 |
1.178 |
| H15 |
-0.873 |
1.896 |
-1.186 |
| H16 |
0.888 |
1.887 |
-1.178 |
| H17 |
-1.206 |
-1.704 |
-1.186 |
| H18 |
-2.079 |
-0.174 |
-1.178 |
| H19 |
2.078 |
-0.192 |
-1.186 |
| H20 |
1.190 |
-1.713 |
-1.178 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5663 | 1.4714 | 1.4714 | 1.4714 | 2.4850 | 2.4850 | 2.4850 | 2.0894 | 2.0885 | 2.0894 | 2.0885 | 2.0894 | 2.0885 | 3.2330 | 3.2260 | 3.2330 | 3.2260 | 3.2330 | 3.2260 |
N2 | 2.5663 | | 2.4850 | 2.4850 | 2.4850 | 1.4714 | 1.4714 | 1.4714 | 3.2330 | 3.2260 | 3.2330 | 3.2260 | 3.2330 | 3.2260 | 2.0894 | 2.0885 | 2.0894 | 2.0885 | 2.0894 | 2.0885 | C3 | 1.4714 | 2.4850 | | 2.3958 | 2.3958 | 1.5627 | 2.8576 | 2.8633 | 1.0927 | 1.0927 | 3.3398 | 2.6302 | 2.6364 | 3.3394 | 2.2111 | 2.2107 | 3.8529 | 3.2513 | 3.2688 | 3.8534 | C4 | 1.4714 | 2.4850 | 2.3958 | | 2.3958 | 2.8576 | 2.8633 | 1.5627 | 2.6364 | 3.3394 | 1.0927 | 1.0927 | 3.3398 | 2.6302 | 3.8529 | 3.2513 | 3.2688 | 3.8534 | 2.2111 | 2.2107 | C5 | 1.4714 | 2.4850 | 2.3958 | 2.3958 | | 2.8633 | 1.5627 | 2.8576 | 3.3398 | 2.6302 | 2.6364 | 3.3394 | 1.0927 | 1.0927 | 3.2688 | 3.8534 | 2.2111 | 2.2107 | 3.8529 | 3.2513 | C6 | 2.4850 | 1.4714 | 1.5627 | 2.8576 | 2.8633 | | 2.3958 | 2.3958 | 2.2111 | 2.2107 | 3.8529 | 3.2513 | 3.2688 | 3.8534 | 1.0927 | 1.0927 | 3.3398 | 2.6302 | 2.6364 | 3.3394 | C7 | 2.4850 | 1.4714 | 2.8576 | 2.8633 | 1.5627 | 2.3958 | | 2.3958 | 3.8529 | 3.2513 | 3.2688 | 3.8534 | 2.2111 | 2.2107 | 2.6364 | 3.3394 | 1.0927 | 1.0927 | 3.3398 | 2.6302 | C8 | 2.4850 | 1.4714 | 2.8633 | 1.5627 | 2.8576 | 2.3958 | 2.3958 | | 3.2688 | 3.8534 | 2.2111 | 2.2107 | 3.8529 | 3.2513 | 3.3398 | 2.6302 | 2.6364 | 3.3394 | 1.0927 | 1.0927 | H9 | 2.0894 | 3.2330 | 1.0927 | 2.6364 | 3.3398 | 2.2111 | 3.8529 | 3.2688 | | 1.7609 | 3.6149 | 2.3959 | 3.6149 | 4.1568 | 2.9449 | 2.3638 | 4.7856 | 4.3108 | 3.3821 | 4.3256 | H10 | 2.0885 | 3.2260 | 1.0927 | 3.3394 | 2.6302 | 2.2107 | 3.2513 | 3.8534 | 1.7609 | | 4.1568 | 3.6127 | 2.3959 | 3.6127 | 2.3638 | 2.9503 | 4.3108 | 3.3491 | 4.3256 | 4.7781 | H11 | 2.0894 | 3.2330 | 3.3398 | 1.0927 | 2.6364 | 3.8529 | 3.2688 | 2.2111 | 3.6149 | 4.1568 | | 1.7609 | 3.6149 | 2.3959 | 4.7856 | 4.3108 | 3.3821 | 4.3256 | 2.9449 | 2.3638 | H12 | 2.0885 | 3.2260 | 2.6302 | 1.0927 | 3.3394 | 3.2513 | 3.8534 | 2.2107 | 2.3959 | 3.6127 | 1.7609 | | 4.1568 | 3.6127 | 4.3108 | 3.3491 | 4.3256 | 4.7781 | 2.3638 | 2.9503 | H13 | 2.0894 | 3.2330 | 2.6364 | 3.3398 | 1.0927 | 3.2688 | 2.2111 | 3.8529 | 3.6149 | 2.3959 | 3.6149 | 4.1568 | | 1.7609 | 3.3821 | 4.3256 | 2.9449 | 2.3638 | 4.7856 | 4.3108 | H14 | 2.0885 | 3.2260 | 3.3394 | 2.6302 | 1.0927 | 3.8534 | 2.2107 | 3.2513 | 4.1568 | 3.6127 | 2.3959 | 3.6127 | 1.7609 | | 4.3256 | 4.7781 | 2.3638 | 2.9503 | 4.3108 | 3.3491 | H15 | 3.2330 | 2.0894 | 2.2111 | 3.8529 | 3.2688 | 1.0927 | 2.6364 | 3.3398 | 2.9449 | 2.3638 | 4.7856 | 4.3108 | 3.3821 | 4.3256 | | 1.7609 | 3.6149 | 2.3959 | 3.6149 | 4.1568 | H16 | 3.2260 | 2.0885 | 2.2107 | 3.2513 | 3.8534 | 1.0927 | 3.3394 | 2.6302 | 2.3638 | 2.9503 | 4.3108 | 3.3491 | 4.3256 | 4.7781 | 1.7609 | | 4.1568 | 3.6127 | 2.3959 | 3.6127 | H17 | 3.2330 | 2.0894 | 3.8529 | 3.2688 | 2.2111 | 3.3398 | 1.0927 | 2.6364 | 4.7856 | 4.3108 | 3.3821 | 4.3256 | 2.9449 | 2.3638 | 3.6149 | 4.1568 | | 1.7609 | 3.6149 | 2.3959 | H18 | 3.2260 | 2.0885 | 3.2513 | 3.8534 | 2.2107 | 2.6302 | 1.0927 | 3.3394 | 4.3108 | 3.3491 | 4.3256 | 4.7781 | 2.3638 | 2.9503 | 2.3959 | 3.6127 | 1.7609 | | 4.1568 | 3.6127 | H19 | 3.2330 | 2.0894 | 3.2688 | 2.2111 | 3.8529 | 2.6364 | 3.3398 | 1.0927 | 3.3821 | 4.3256 | 2.9449 | 2.3638 | 4.7856 | 4.3108 | 3.6149 | 2.3959 | 3.6149 | 4.1568 | | 1.7609 | H20 | 3.2260 | 2.0885 | 3.8534 | 2.2107 | 3.2513 | 3.3394 | 2.6302 | 1.0927 | 4.3256 | 4.7781 | 2.3638 | 2.9503 | 4.3108 | 3.3491 | 4.1568 | 3.6127 | 2.3959 | 3.6127 | 1.7609 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.937 |
|
N1 |
C3 |
H9 |
108.236 |
| N1 |
C3 |
H10 |
108.166 |
|
N1 |
C4 |
C8 |
109.937 |
| N1 |
C4 |
H11 |
108.236 |
|
N1 |
C4 |
H12 |
108.166 |
| N1 |
C5 |
C7 |
109.937 |
|
N1 |
C5 |
H13 |
108.236 |
| N1 |
C5 |
H14 |
108.166 |
|
N2 |
C6 |
C3 |
109.937 |
| N2 |
C6 |
H15 |
108.236 |
|
N2 |
C6 |
H16 |
108.166 |
| N2 |
C7 |
C5 |
109.937 |
|
N2 |
C7 |
H17 |
108.236 |
| N2 |
C7 |
H18 |
108.166 |
|
N2 |
C8 |
C4 |
109.937 |
| N2 |
C8 |
H19 |
108.236 |
|
N2 |
C8 |
H20 |
108.166 |
| C3 |
N1 |
C4 |
109.000 |
|
C3 |
N1 |
C5 |
109.000 |
| C3 |
C6 |
H15 |
111.516 |
|
C3 |
C6 |
H16 |
111.488 |
| C4 |
N1 |
C5 |
109.000 |
|
C4 |
C8 |
H19 |
111.516 |
| C4 |
C8 |
H20 |
111.488 |
|
C5 |
C6 |
H15 |
101.923 |
| C5 |
C6 |
H16 |
150.689 |
|
C6 |
N2 |
C7 |
109.000 |
| C6 |
N2 |
C8 |
109.000 |
|
C6 |
C3 |
H9 |
111.516 |
| C6 |
C3 |
H10 |
111.488 |
|
C7 |
N2 |
C8 |
109.000 |
| C7 |
C5 |
H13 |
111.516 |
|
C7 |
C5 |
H14 |
111.488 |
| C8 |
C4 |
H11 |
111.516 |
|
C8 |
C4 |
H12 |
111.488 |
| H9 |
C3 |
H10 |
107.364 |
|
H11 |
C4 |
H12 |
107.364 |
| H13 |
C5 |
H14 |
107.364 |
|
H15 |
C6 |
H16 |
107.364 |
| H17 |
C7 |
H18 |
107.364 |
|
H19 |
C8 |
H20 |
107.364 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.117 |
|
|
|
| 2 |
N |
-0.117 |
|
|
|
| 3 |
C |
-0.224 |
|
|
|
| 4 |
C |
-0.224 |
|
|
|
| 5 |
C |
-0.224 |
|
|
|
| 6 |
C |
-0.224 |
|
|
|
| 7 |
C |
-0.224 |
|
|
|
| 8 |
C |
-0.224 |
|
|
|
| 9 |
H |
0.131 |
|
|
|
| 10 |
H |
0.131 |
|
|
|
| 11 |
H |
0.131 |
|
|
|
| 12 |
H |
0.131 |
|
|
|
| 13 |
H |
0.131 |
|
|
|
| 14 |
H |
0.131 |
|
|
|
| 15 |
H |
0.131 |
|
|
|
| 16 |
H |
0.131 |
|
|
|
| 17 |
H |
0.131 |
|
|
|
| 18 |
H |
0.131 |
|
|
|
| 19 |
H |
0.131 |
|
|
|
| 20 |
H |
0.131 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.396 |
0.000 |
0.000 |
| y |
0.000 |
-47.396 |
0.000 |
| z |
0.000 |
0.000 |
-58.908 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.756 |
0.000 |
0.000 |
| y |
0.000 |
5.756 |
0.000 |
| z |
0.000 |
0.000 |
-11.513 |
|
| Polar |
|---|
| 3z2-r2 | -23.025 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.121 |
0.000 |
0.000 |
| y |
0.000 |
12.122 |
0.000 |
| z |
0.000 |
0.000 |
11.129 |
<r2> (average value of r
2) Å
2
| <r2> |
214.909 |
| (<r2>)1/2 |
14.660 |