Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3723 |
3575 |
28.02 |
|
|
|
| 2 |
A |
3594 |
3452 |
28.87 |
|
|
|
| 3 |
A |
3149 |
3024 |
11.47 |
|
|
|
| 4 |
A |
3137 |
3013 |
16.78 |
|
|
|
| 5 |
A |
3066 |
2945 |
8.05 |
|
|
|
| 6 |
A |
1814 |
1742 |
312.14 |
|
|
|
| 7 |
A |
1647 |
1582 |
98.44 |
|
|
|
| 8 |
A |
1520 |
1459 |
8.01 |
|
|
|
| 9 |
A |
1503 |
1444 |
5.67 |
|
|
|
| 10 |
A |
1427 |
1370 |
45.91 |
|
|
|
| 11 |
A |
1360 |
1306 |
129.85 |
|
|
|
| 12 |
A |
1134 |
1089 |
0.37 |
|
|
|
| 13 |
A |
1069 |
1027 |
7.84 |
|
|
|
| 14 |
A |
995 |
955 |
7.72 |
|
|
|
| 15 |
A |
846 |
812 |
2.24 |
|
|
|
| 16 |
A |
675 |
648 |
17.25 |
|
|
|
| 17 |
A |
543 |
522 |
14.78 |
|
|
|
| 18 |
A |
520 |
500 |
7.21 |
|
|
|
| 19 |
A |
430 |
413 |
4.42 |
|
|
|
| 20 |
A |
134 |
129 |
235.04 |
|
|
|
| 21 |
A |
38 |
36 |
11.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16162.5 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 15520.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
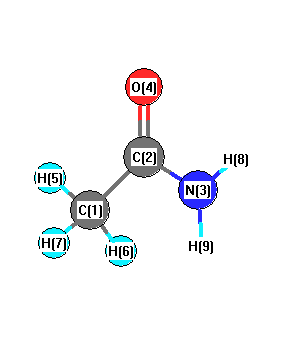
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.530 |
-0.357 |
|
-0.534 |
| 2 |
C |
0.579 |
0.788 |
|
0.797 |
| 3 |
N |
-0.739 |
-0.953 |
|
-0.957 |
| 4 |
O |
-0.495 |
-0.569 |
|
-0.555 |
| 5 |
H |
0.188 |
0.105 |
|
0.155 |
| 6 |
H |
0.141 |
0.086 |
|
0.134 |
| 7 |
H |
0.185 |
0.097 |
|
0.147 |
| 8 |
H |
0.340 |
0.407 |
|
0.408 |
| 9 |
H |
0.332 |
0.395 |
|
0.405 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.069 |
-3.699 |
0.277 |
3.710 |
| CHELPG |
-0.074 |
-3.736 |
0.286 |
3.748 |
| AIM |
|
|
|
|
| ESP |
-0.092 |
-3.721 |
0.294 |
3.734 |
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
75.300 |
| (<r2>)1/2 |
8.678 |