Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3277 |
3147 |
1.49 |
|
|
|
| 2 |
A |
3245 |
3116 |
2.13 |
|
|
|
| 3 |
A |
3139 |
3014 |
15.97 |
|
|
|
| 4 |
A |
3110 |
2987 |
15.11 |
|
|
|
| 5 |
A |
3055 |
2933 |
23.07 |
|
|
|
| 6 |
A |
1586 |
1523 |
28.09 |
|
|
|
| 7 |
A |
1514 |
1454 |
29.69 |
|
|
|
| 8 |
A |
1507 |
1447 |
5.68 |
|
|
|
| 9 |
A |
1495 |
1436 |
25.98 |
|
|
|
| 10 |
A |
1442 |
1385 |
1.40 |
|
|
|
| 11 |
A |
1360 |
1306 |
13.40 |
|
|
|
| 12 |
A |
1266 |
1216 |
1.64 |
|
|
|
| 13 |
A |
1174 |
1127 |
4.58 |
|
|
|
| 14 |
A |
1080 |
1037 |
4.39 |
|
|
|
| 15 |
A |
1028 |
988 |
8.66 |
|
|
|
| 16 |
A |
950 |
912 |
16.53 |
|
|
|
| 17 |
A |
881 |
846 |
30.14 |
|
|
|
| 18 |
A |
824 |
791 |
11.25 |
|
|
|
| 19 |
A |
817 |
785 |
37.99 |
|
|
|
| 20 |
A |
735 |
705 |
7.66 |
|
|
|
| 21 |
A |
668 |
642 |
2.34 |
|
|
|
| 22 |
A |
652 |
626 |
0.51 |
|
|
|
| 23 |
A |
556 |
534 |
2.50 |
|
|
|
| 24 |
A |
487 |
468 |
1.91 |
|
|
|
| 25 |
A |
333 |
320 |
2.25 |
|
|
|
| 26 |
A |
232 |
223 |
3.45 |
|
|
|
| 27 |
A |
129 |
124 |
0.44 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18271.2 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 17545.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
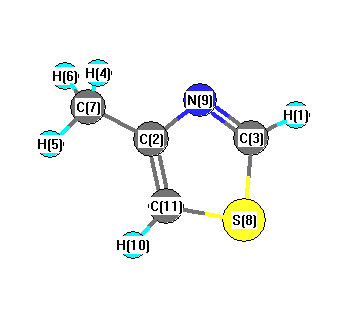
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.182 |
0.109 |
|
0.152 |
| 2 |
C |
0.308 |
0.474 |
|
0.563 |
| 3 |
C |
-0.128 |
0.156 |
|
0.076 |
| 4 |
H |
0.172 |
0.135 |
|
0.181 |
| 5 |
H |
0.152 |
0.094 |
|
0.141 |
| 6 |
H |
0.172 |
0.136 |
|
0.181 |
| 7 |
C |
-0.503 |
-0.446 |
|
-0.624 |
| 8 |
S |
0.237 |
-0.024 |
|
0.001 |
| 9 |
N |
-0.381 |
-0.493 |
|
-0.485 |
| 10 |
H |
0.176 |
0.206 |
|
0.235 |
| 11 |
C |
-0.386 |
-0.348 |
|
-0.420 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.096 |
-1.135 |
0.000 |
1.139 |
| CHELPG |
0.175 |
-1.105 |
0.000 |
1.119 |
| AIM |
|
|
|
|
| ESP |
0.106 |
-1.080 |
0.000 |
1.085 |
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
180.274 |
| (<r2>)1/2 |
13.427 |