Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3645 |
3500 |
35.29 |
|
|
|
| 2 |
A |
3293 |
3163 |
4.65 |
|
|
|
| 3 |
A |
3263 |
3133 |
8.45 |
|
|
|
| 4 |
A |
3161 |
3035 |
4.94 |
|
|
|
| 5 |
A |
3089 |
2966 |
22.89 |
|
|
|
| 6 |
A |
3042 |
2921 |
35.41 |
|
|
|
| 7 |
A |
1609 |
1545 |
35.35 |
|
|
|
| 8 |
A |
1538 |
1477 |
2.75 |
|
|
|
| 9 |
A |
1516 |
1455 |
3.45 |
|
|
|
| 10 |
A |
1514 |
1454 |
7.43 |
|
|
|
| 11 |
A |
1457 |
1399 |
42.41 |
|
|
|
| 12 |
A |
1441 |
1383 |
0.42 |
|
|
|
| 13 |
A |
1404 |
1348 |
1.34 |
|
|
|
| 14 |
A |
1284 |
1233 |
15.60 |
|
|
|
| 15 |
A |
1192 |
1145 |
4.10 |
|
|
|
| 16 |
A |
1144 |
1099 |
1.96 |
|
|
|
| 17 |
A |
1109 |
1065 |
24.43 |
|
|
|
| 18 |
A |
1075 |
1032 |
1.72 |
|
|
|
| 19 |
A |
1015 |
974 |
9.98 |
|
|
|
| 20 |
A |
974 |
935 |
1.43 |
|
|
|
| 21 |
A |
928 |
891 |
3.08 |
|
|
|
| 22 |
A |
849 |
816 |
9.81 |
|
|
|
| 23 |
A |
722 |
693 |
31.20 |
|
|
|
| 24 |
A |
688 |
661 |
6.13 |
|
|
|
| 25 |
A |
679 |
652 |
1.35 |
|
|
|
| 26 |
A |
642 |
616 |
15.34 |
|
|
|
| 27 |
A |
501 |
481 |
77.27 |
|
|
|
| 28 |
A |
346 |
333 |
5.06 |
|
|
|
| 29 |
A |
251 |
241 |
5.89 |
|
|
|
| 30 |
A |
83 |
80 |
0.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21726.3 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 20863.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
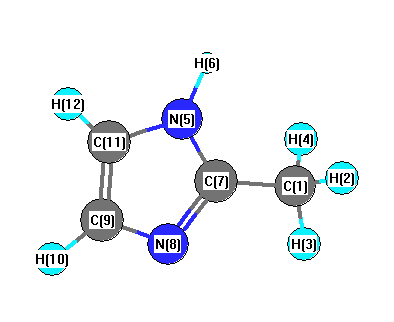
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.530 |
-0.262 |
-0.010 |
-0.457 |
| 2 |
H |
0.160 |
0.063 |
0.023 |
0.111 |
| 3 |
H |
0.192 |
0.100 |
0.059 |
0.149 |
| 4 |
H |
0.160 |
0.063 |
0.023 |
0.111 |
| 5 |
N |
-0.589 |
-0.277 |
-1.280 |
-0.313 |
| 6 |
H |
0.328 |
0.303 |
0.423 |
0.321 |
| 7 |
C |
0.481 |
0.446 |
1.053 |
0.549 |
| 8 |
N |
-0.466 |
-0.554 |
-1.180 |
-0.564 |
| 9 |
C |
-0.033 |
0.151 |
0.420 |
0.106 |
| 10 |
H |
0.130 |
0.072 |
0.058 |
0.104 |
| 11 |
C |
0.019 |
-0.275 |
0.336 |
-0.318 |
| 12 |
H |
0.147 |
0.170 |
0.073 |
0.200 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.482 |
3.550 |
-0.001 |
3.582 |
| CHELPG |
0.423 |
3.509 |
-0.001 |
3.535 |
| AIM |
0.351 |
0.121 |
0.000 |
0.371 |
| ESP |
0.467 |
3.524 |
-0.001 |
3.555 |
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
139.204 |
| (<r2>)1/2 |
11.798 |