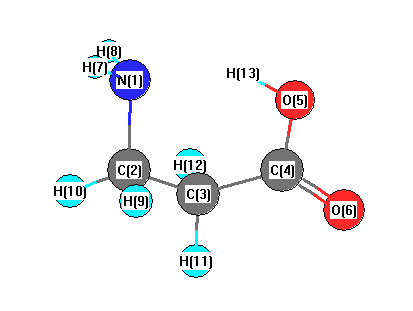
Vibrational Frequencies calculated at mPW1PW91/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3630 |
3443 |
7.81 |
|
|
|
| 2 |
A |
3538 |
3355 |
1.95 |
|
|
|
| 3 |
A |
3216 |
3050 |
740.52 |
|
|
|
| 4 |
A |
3170 |
3006 |
5.93 |
|
|
|
| 5 |
A |
3128 |
2967 |
20.72 |
|
|
|
| 6 |
A |
3070 |
2911 |
7.32 |
|
|
|
| 7 |
A |
3054 |
2896 |
72.56 |
|
|
|
| 8 |
A |
1901 |
1803 |
385.90 |
|
|
|
| 9 |
A |
1700 |
1612 |
36.73 |
|
|
|
| 10 |
A |
1547 |
1467 |
92.16 |
|
|
|
| 11 |
A |
1535 |
1455 |
210.30 |
|
|
|
| 12 |
A |
1494 |
1417 |
8.73 |
|
|
|
| 13 |
A |
1441 |
1367 |
10.70 |
|
|
|
| 14 |
A |
1391 |
1319 |
11.47 |
|
|
|
| 15 |
A |
1337 |
1268 |
4.73 |
|
|
|
| 16 |
A |
1322 |
1254 |
27.47 |
|
|
|
| 17 |
A |
1278 |
1212 |
57.60 |
|
|
|
| 18 |
A |
1166 |
1106 |
5.76 |
|
|
|
| 19 |
A |
1107 |
1050 |
6.02 |
|
|
|
| 20 |
A |
1044 |
990 |
11.58 |
|
|
|
| 21 |
A |
1020 |
968 |
109.11 |
|
|
|
| 22 |
A |
974 |
924 |
13.53 |
|
|
|
| 23 |
A |
933 |
885 |
26.94 |
|
|
|
| 24 |
A |
887 |
841 |
40.24 |
|
|
|
| 25 |
A |
827 |
784 |
22.24 |
|
|
|
| 26 |
A |
703 |
666 |
10.99 |
|
|
|
| 27 |
A |
583 |
553 |
2.97 |
|
|
|
| 28 |
A |
495 |
470 |
10.18 |
|
|
|
| 29 |
A |
415 |
393 |
14.40 |
|
|
|
| 30 |
A |
338 |
321 |
7.72 |
|
|
|
| 31 |
A |
299 |
283 |
8.98 |
|
|
|
| 32 |
A |
205 |
194 |
3.89 |
|
|
|
| 33 |
A |
91 |
86 |
0.39 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24419.4 cm
-1
Scaled (by 0.9483) Zero Point Vibrational Energy (zpe) 23156.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.810 |
|
|
|
| 2 |
C |
-0.225 |
|
|
|
| 3 |
C |
-0.453 |
|
|
|
| 4 |
C |
0.614 |
|
|
|
| 5 |
O |
-0.611 |
|
|
|
| 6 |
O |
-0.473 |
|
|
|
| 7 |
H |
0.352 |
|
|
|
| 8 |
H |
0.345 |
|
|
|
| 9 |
H |
0.180 |
|
|
|
| 10 |
H |
0.202 |
|
|
|
| 11 |
H |
0.215 |
|
|
|
| 12 |
H |
0.199 |
|
|
|
| 13 |
H |
0.466 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
6.622 |
-1.243 |
0.602 |
6.764 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.070 |
1.749 |
-0.234 |
| y |
1.749 |
-36.811 |
0.086 |
| z |
-0.234 |
0.086 |
-33.001 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.164 |
1.749 |
-0.234 |
| y |
1.749 |
-1.276 |
0.086 |
| z |
-0.234 |
0.086 |
4.440 |
|
| Polar |
|---|
| 3z2-r2 | 8.879 |
|---|
| x2-y2 | -1.259 |
|---|
| xy | 1.749 |
|---|
| xz | -0.234 |
|---|
| yz | 0.086 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.803 |
0.448 |
0.117 |
| y |
0.448 |
6.288 |
-0.016 |
| z |
0.117 |
-0.016 |
5.107 |
<r2> (average value of r
2) Å
2
| <r2> |
170.472 |
| (<r2>)1/2 |
13.056 |