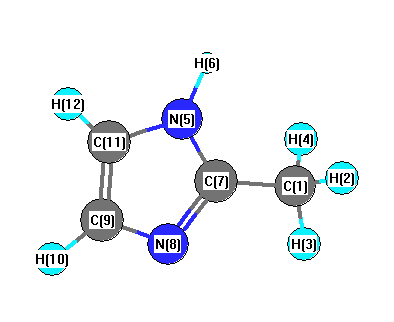
Vibrational Frequencies calculated at mPW1PW91/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3697 |
3506 |
44.91 |
|
|
|
| 2 |
A |
3324 |
3152 |
3.40 |
|
|
|
| 3 |
A |
3292 |
3122 |
7.54 |
|
|
|
| 4 |
A |
3198 |
3033 |
3.17 |
|
|
|
| 5 |
A |
3131 |
2969 |
18.16 |
|
|
|
| 6 |
A |
3072 |
2914 |
31.13 |
|
|
|
| 7 |
A |
1636 |
1551 |
39.57 |
|
|
|
| 8 |
A |
1557 |
1476 |
4.50 |
|
|
|
| 9 |
A |
1532 |
1452 |
2.67 |
|
|
|
| 10 |
A |
1517 |
1438 |
8.95 |
|
|
|
| 11 |
A |
1479 |
1402 |
43.26 |
|
|
|
| 12 |
A |
1449 |
1374 |
0.26 |
|
|
|
| 13 |
A |
1424 |
1351 |
1.12 |
|
|
|
| 14 |
A |
1304 |
1236 |
14.88 |
|
|
|
| 15 |
A |
1219 |
1156 |
2.89 |
|
|
|
| 16 |
A |
1156 |
1096 |
4.64 |
|
|
|
| 17 |
A |
1127 |
1068 |
27.52 |
|
|
|
| 18 |
A |
1076 |
1021 |
2.33 |
|
|
|
| 19 |
A |
1022 |
969 |
8.39 |
|
|
|
| 20 |
A |
983 |
932 |
1.16 |
|
|
|
| 21 |
A |
934 |
886 |
3.03 |
|
|
|
| 22 |
A |
864 |
819 |
10.37 |
|
|
|
| 23 |
A |
732 |
694 |
38.58 |
|
|
|
| 24 |
A |
697 |
661 |
5.54 |
|
|
|
| 25 |
A |
689 |
653 |
1.28 |
|
|
|
| 26 |
A |
650 |
616 |
16.49 |
|
|
|
| 27 |
A |
525 |
498 |
80.56 |
|
|
|
| 28 |
A |
348 |
330 |
5.34 |
|
|
|
| 29 |
A |
254 |
241 |
6.26 |
|
|
|
| 30 |
A |
83 |
79 |
0.23 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21984.8 cm
-1
Scaled (by 0.9483) Zero Point Vibrational Energy (zpe) 20848.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.604 |
|
|
|
| 2 |
H |
0.188 |
|
|
|
| 3 |
H |
0.221 |
|
|
|
| 4 |
H |
0.187 |
|
|
|
| 5 |
N |
-0.631 |
|
|
|
| 6 |
H |
0.354 |
|
|
|
| 7 |
C |
0.490 |
|
|
|
| 8 |
N |
-0.481 |
|
|
|
| 9 |
C |
-0.067 |
|
|
|
| 10 |
H |
0.167 |
|
|
|
| 11 |
C |
-0.009 |
|
|
|
| 12 |
H |
0.184 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.498 |
3.605 |
-0.002 |
3.639 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.022 |
0.521 |
0.001 |
| y |
0.521 |
-33.233 |
-0.006 |
| z |
0.001 |
-0.006 |
-37.278 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.233 |
0.521 |
0.001 |
| y |
0.521 |
0.417 |
-0.006 |
| z |
0.001 |
-0.006 |
-5.650 |
|
| Polar |
|---|
| 3z2-r2 | -11.300 |
|---|
| x2-y2 | 3.211 |
|---|
| xy | 0.521 |
|---|
| xz | 0.001 |
|---|
| yz | -0.006 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
138.057 |
| (<r2>)1/2 |
11.750 |