Vibrational Frequencies calculated at B3LYP/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3142 |
3040 |
6.48 |
|
|
|
| 2 |
A |
3084 |
2984 |
5.22 |
|
|
|
| 3 |
A |
3069 |
2969 |
2.50 |
|
|
|
| 4 |
A |
3030 |
2932 |
0.56 |
|
|
|
| 5 |
A |
1790 |
1732 |
21.25 |
|
|
|
| 6 |
A |
1481 |
1432 |
6.79 |
|
|
|
| 7 |
A |
1474 |
1426 |
15.75 |
|
|
|
| 8 |
A |
1462 |
1414 |
5.17 |
|
|
|
| 9 |
A |
1390 |
1345 |
3.85 |
|
|
|
| 10 |
A |
1271 |
1229 |
21.27 |
|
|
|
| 11 |
A |
1144 |
1107 |
1.63 |
|
|
|
| 12 |
A |
1080 |
1045 |
0.66 |
|
|
|
| 13 |
A |
937 |
907 |
1.76 |
|
|
|
| 14 |
A |
784 |
759 |
0.13 |
|
|
|
| 15 |
A |
628 |
607 |
1.52 |
|
|
|
| 16 |
A |
488 |
472 |
6.11 |
|
|
|
| 17 |
A |
323 |
312 |
0.84 |
|
|
|
| 18 |
A |
164 |
159 |
0.01 |
|
|
|
| 19 |
A |
144 |
140 |
1.03 |
|
|
|
| 20 |
A |
56 |
54 |
8.52 |
|
|
|
| 21 |
B |
3142 |
3040 |
8.55 |
|
|
|
| 22 |
B |
3132 |
3030 |
5.68 |
|
|
|
| 23 |
B |
3084 |
2984 |
0.22 |
|
|
|
| 24 |
B |
3030 |
2932 |
2.17 |
|
|
|
| 25 |
B |
1760 |
1703 |
383.00 |
|
|
|
| 26 |
B |
1474 |
1426 |
0.64 |
|
|
|
| 27 |
B |
1466 |
1419 |
28.06 |
|
|
|
| 28 |
B |
1389 |
1344 |
77.60 |
|
|
|
| 29 |
B |
1256 |
1215 |
85.66 |
|
|
|
| 30 |
B |
1186 |
1148 |
198.76 |
|
|
|
| 31 |
B |
1063 |
1029 |
9.81 |
|
|
|
| 32 |
B |
1001 |
968 |
1.33 |
|
|
|
| 33 |
B |
892 |
863 |
32.32 |
|
|
|
| 34 |
B |
797 |
771 |
13.90 |
|
|
|
| 35 |
B |
550 |
532 |
26.38 |
|
|
|
| 36 |
B |
501 |
485 |
1.37 |
|
|
|
| 37 |
B |
411 |
398 |
2.49 |
|
|
|
| 38 |
B |
163 |
158 |
0.05 |
|
|
|
| 39 |
B |
47 |
46 |
11.98 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 26643.9 cm
-1
Scaled (by 0.9675) Zero Point Vibrational Energy (zpe) 25778.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
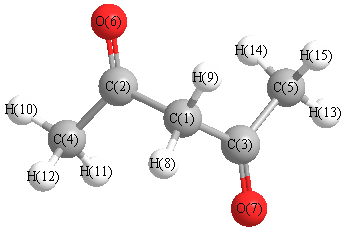
Charges from optimized geometry at B3LYP/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.006 |
|
|
|
| 2 |
C |
0.633 |
|
|
|
| 3 |
C |
0.633 |
|
|
|
| 4 |
C |
-0.637 |
|
|
|
| 5 |
C |
-0.637 |
|
|
|
| 6 |
O |
-0.793 |
|
|
|
| 7 |
O |
-0.793 |
|
|
|
| 8 |
H |
0.146 |
|
|
|
| 9 |
H |
0.146 |
|
|
|
| 10 |
H |
0.200 |
|
|
|
| 11 |
H |
0.227 |
|
|
|
| 12 |
H |
0.221 |
|
|
|
| 13 |
H |
0.200 |
|
|
|
| 14 |
H |
0.227 |
|
|
|
| 15 |
H |
0.221 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.576 |
1.576 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-44.536 |
-10.061 |
0.000 |
| y |
-10.061 |
-46.612 |
0.000 |
| z |
0.000 |
0.000 |
-40.605 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.928 |
-10.061 |
0.000 |
| y |
-10.061 |
-4.041 |
0.000 |
| z |
0.000 |
0.000 |
4.969 |
|
| Polar |
|---|
| 3z2-r2 | 9.938 |
|---|
| x2-y2 | 2.076 |
|---|
| xy | -10.061 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.912 |
-0.349 |
0.000 |
| y |
-0.349 |
11.207 |
0.000 |
| z |
0.000 |
0.000 |
8.420 |
<r2> (average value of r
2) Å
2
| <r2> |
232.272 |
| (<r2>)1/2 |
15.240 |