Jump to
S1C2
Energy calculated at BLYP/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -342.333248 |
| Energy at 298.15K | |
| HF Energy | -342.333248 |
| Nuclear repulsion energy | 241.909274 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2956 |
2961 |
65.32 |
|
|
|
| 2 |
A1 |
1856 |
1859 |
512.15 |
|
|
|
| 3 |
A1 |
1474 |
1476 |
2.97 |
|
|
|
| 4 |
A1 |
1330 |
1332 |
1.32 |
|
|
|
| 5 |
A1 |
1109 |
1111 |
200.74 |
|
|
|
| 6 |
A1 |
908 |
910 |
9.59 |
|
|
|
| 7 |
A1 |
774 |
775 |
33.44 |
|
|
|
| 8 |
A1 |
690 |
691 |
0.76 |
|
|
|
| 9 |
A2 |
2976 |
2981 |
0.00 |
|
|
|
| 10 |
A2 |
1193 |
1195 |
0.00 |
|
|
|
| 11 |
A2 |
1119 |
1121 |
0.00 |
|
|
|
| 12 |
A2 |
156i |
157i |
0.00 |
|
|
|
| 13 |
B1 |
2999 |
3004 |
59.40 |
|
|
|
| 14 |
B1 |
1207 |
1209 |
0.97 |
|
|
|
| 15 |
B1 |
826 |
827 |
0.27 |
|
|
|
| 16 |
B1 |
704 |
706 |
13.17 |
|
|
|
| 17 |
B1 |
146 |
146 |
1.37 |
|
|
|
| 18 |
B2 |
2948 |
2953 |
35.15 |
|
|
|
| 19 |
B2 |
1463 |
1465 |
2.01 |
|
|
|
| 20 |
B2 |
1346 |
1348 |
17.24 |
|
|
|
| 21 |
B2 |
1103 |
1104 |
0.00 |
|
|
|
| 22 |
B2 |
967 |
968 |
268.49 |
|
|
|
| 23 |
B2 |
714 |
715 |
7.90 |
|
|
|
| 24 |
B2 |
477 |
477 |
0.46 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 15563.6 cm
-1
Scaled (by 1.0016) Zero Point Vibrational Energy (zpe) 15588.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/cc-pVDZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.879 |
| O2 |
0.000 |
0.000 |
2.078 |
| O3 |
0.000 |
1.152 |
0.060 |
| O4 |
0.000 |
-1.152 |
0.060 |
| C5 |
0.000 |
0.783 |
-1.305 |
| C6 |
0.000 |
-0.783 |
-1.305 |
| H7 |
-0.901 |
1.203 |
-1.799 |
| H8 |
0.901 |
1.203 |
-1.799 |
| H9 |
0.901 |
-1.203 |
-1.799 |
| H10 |
-0.901 |
-1.203 |
-1.799 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1991 | 1.4136 | 1.4136 | 2.3198 | 2.3198 | 3.0705 | 3.0705 | 3.0705 | 3.0705 |
O2 | 1.1991 | | 2.3239 | 2.3239 | 3.4722 | 3.4722 | 4.1577 | 4.1577 | 4.1577 | 4.1577 | O3 | 1.4136 | 2.3239 | | 2.3039 | 1.4136 | 2.3673 | 2.0656 | 2.0656 | 3.1322 | 3.1322 | O4 | 1.4136 | 2.3239 | 2.3039 | | 2.3673 | 1.4136 | 3.1322 | 3.1322 | 2.0656 | 2.0656 | C5 | 2.3198 | 3.4722 | 1.4136 | 2.3673 | | 1.5653 | 1.1098 | 1.1098 | 2.2357 | 2.2357 | C6 | 2.3198 | 3.4722 | 2.3673 | 1.4136 | 1.5653 | | 2.2357 | 2.2357 | 1.1098 | 1.1098 | H7 | 3.0705 | 4.1577 | 2.0656 | 3.1322 | 1.1098 | 2.2357 | | 1.8011 | 3.0057 | 2.4063 | H8 | 3.0705 | 4.1577 | 2.0656 | 3.1322 | 1.1098 | 2.2357 | 1.8011 | | 2.4063 | 3.0057 | H9 | 3.0705 | 4.1577 | 3.1322 | 2.0656 | 2.2357 | 1.1098 | 3.0057 | 2.4063 | | 1.8011 | H10 | 3.0705 | 4.1577 | 3.1322 | 2.0656 | 2.2357 | 1.1098 | 2.4063 | 3.0057 | 1.8011 | |
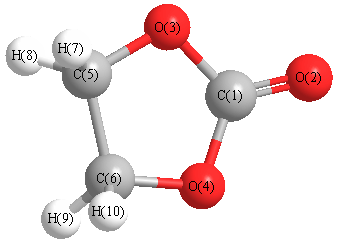 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
110.276 |
|
C1 |
O4 |
C6 |
110.276 |
| O2 |
C1 |
O3 |
125.421 |
|
O2 |
C1 |
O4 |
125.421 |
| O3 |
C1 |
O4 |
109.158 |
|
O3 |
C5 |
C6 |
105.144 |
| O3 |
C5 |
H7 |
109.298 |
|
O3 |
C5 |
H8 |
109.298 |
| O4 |
C6 |
C5 |
105.144 |
|
O4 |
C6 |
H9 |
109.298 |
| O4 |
C6 |
H10 |
109.298 |
|
C5 |
C6 |
H9 |
112.266 |
| C5 |
C6 |
H10 |
112.266 |
|
C6 |
C5 |
H7 |
112.266 |
| C6 |
C5 |
H8 |
112.266 |
|
H7 |
C5 |
H8 |
108.478 |
| H9 |
C6 |
H10 |
108.478 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.246 |
|
|
|
| 2 |
O |
-0.165 |
|
|
|
| 3 |
O |
-0.217 |
|
|
|
| 4 |
O |
-0.217 |
|
|
|
| 5 |
C |
0.131 |
|
|
|
| 6 |
C |
0.131 |
|
|
|
| 7 |
H |
0.023 |
|
|
|
| 8 |
H |
0.023 |
|
|
|
| 9 |
H |
0.023 |
|
|
|
| 10 |
H |
0.023 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.403 |
4.403 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.197 |
0.000 |
0.000 |
| y |
0.000 |
-35.924 |
0.000 |
| z |
0.000 |
0.000 |
-35.417 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.474 |
0.000 |
0.000 |
| y |
0.000 |
-2.117 |
0.000 |
| z |
0.000 |
0.000 |
-1.357 |
|
| Polar |
|---|
| 3z2-r2 | -2.714 |
|---|
| x2-y2 | 3.727 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.368 |
0.000 |
0.000 |
| y |
0.000 |
5.447 |
0.000 |
| z |
0.000 |
0.000 |
7.800 |
<r2> (average value of r
2) Å
2
| <r2> |
131.779 |
| (<r2>)1/2 |
11.479 |
Jump to
S1C1
Energy calculated at BLYP/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -342.333806 |
| Energy at 298.15K | -342.339997 |
| HF Energy | -342.333806 |
| Nuclear repulsion energy | 242.381526 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3010 |
3014 |
23.81 |
|
|
|
| 2 |
A |
2930 |
2935 |
33.40 |
|
|
|
| 3 |
A |
1859 |
1862 |
500.35 |
|
|
|
| 4 |
A |
1463 |
1465 |
3.17 |
|
|
|
| 5 |
A |
1337 |
1339 |
5.64 |
|
|
|
| 6 |
A |
1198 |
1200 |
11.25 |
|
|
|
| 7 |
A |
1129 |
1131 |
46.57 |
|
|
|
| 8 |
A |
1085 |
1087 |
135.82 |
|
|
|
| 9 |
A |
913 |
914 |
5.41 |
|
|
|
| 10 |
A |
768 |
770 |
36.53 |
|
|
|
| 11 |
A |
688 |
689 |
0.97 |
|
|
|
| 12 |
A |
124 |
124 |
0.33 |
|
|
|
| 13 |
B |
3019 |
3024 |
35.87 |
|
|
|
| 14 |
B |
2936 |
2940 |
63.46 |
|
|
|
| 15 |
B |
1456 |
1458 |
2.82 |
|
|
|
| 16 |
B |
1341 |
1343 |
14.99 |
|
|
|
| 17 |
B |
1207 |
1209 |
3.00 |
|
|
|
| 18 |
B |
1086 |
1087 |
3.56 |
|
|
|
| 19 |
B |
964 |
966 |
232.12 |
|
|
|
| 20 |
B |
851 |
852 |
24.93 |
|
|
|
| 21 |
B |
712 |
713 |
14.15 |
|
|
|
| 22 |
B |
662 |
663 |
3.31 |
|
|
|
| 23 |
B |
476 |
477 |
0.65 |
|
|
|
| 24 |
B |
153 |
154 |
1.28 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 15682.8 cm
-1
Scaled (by 1.0016) Zero Point Vibrational Energy (zpe) 15707.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/cc-pVDZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.875 |
| O2 |
0.000 |
0.000 |
2.074 |
| O3 |
0.000 |
1.152 |
0.050 |
| O4 |
0.000 |
-1.152 |
0.050 |
| C5 |
0.186 |
0.753 |
-1.296 |
| C6 |
-0.186 |
-0.753 |
-1.296 |
| H7 |
-0.468 |
1.360 |
-1.952 |
| H8 |
1.246 |
0.919 |
-1.591 |
| H9 |
0.468 |
-1.360 |
-1.952 |
| H10 |
-1.246 |
-0.919 |
-1.591 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1987 | 1.4169 | 1.4169 | 2.3056 | 2.3056 | 3.1714 | 2.9115 | 3.1714 | 2.9115 |
O2 | 1.1987 | | 2.3284 | 2.3284 | 3.4581 | 3.4581 | 4.2745 | 3.9780 | 4.2745 | 3.9780 | O3 | 1.4169 | 2.3284 | | 2.3046 | 1.4169 | 2.3407 | 2.0663 | 2.0737 | 3.2465 | 2.9219 | O4 | 1.4169 | 2.3284 | 2.3046 | | 2.3407 | 1.4169 | 3.2465 | 2.9219 | 2.0663 | 2.0737 | C5 | 2.3056 | 3.4581 | 1.4169 | 2.3407 | | 1.5516 | 1.1071 | 1.1127 | 2.2306 | 2.2217 | C6 | 2.3056 | 3.4581 | 2.3407 | 1.4169 | 1.5516 | | 2.2306 | 2.2217 | 1.1071 | 1.1127 | H7 | 3.1714 | 4.2745 | 2.0663 | 3.2465 | 1.1071 | 2.2306 | | 1.8063 | 2.8771 | 2.4360 | H8 | 2.9115 | 3.9780 | 2.0737 | 2.9219 | 1.1127 | 2.2217 | 1.8063 | | 2.4360 | 3.0975 | H9 | 3.1714 | 4.2745 | 3.2465 | 2.0663 | 2.2306 | 1.1071 | 2.8771 | 2.4360 | | 1.8063 | H10 | 2.9115 | 3.9780 | 2.9219 | 2.0737 | 2.2217 | 1.1127 | 2.4360 | 3.0975 | 1.8063 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
108.903 |
|
C1 |
O4 |
C6 |
108.903 |
| O2 |
C1 |
O3 |
125.585 |
|
O2 |
C1 |
O4 |
125.585 |
| O3 |
C1 |
O4 |
108.829 |
|
O3 |
C5 |
C6 |
104.002 |
| O3 |
C5 |
H7 |
109.288 |
|
O3 |
C5 |
H8 |
109.538 |
| O4 |
C6 |
C5 |
104.002 |
|
O4 |
C6 |
H9 |
109.288 |
| O4 |
C6 |
H10 |
109.538 |
|
C5 |
C6 |
H9 |
113.002 |
| C5 |
C6 |
H10 |
111.944 |
|
C6 |
C5 |
H7 |
113.002 |
| C6 |
C5 |
H8 |
111.944 |
|
H7 |
C5 |
H8 |
108.923 |
| H9 |
C6 |
H10 |
108.923 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.245 |
|
|
|
| 2 |
O |
-0.162 |
|
|
|
| 3 |
O |
-0.220 |
|
|
|
| 4 |
O |
-0.220 |
|
|
|
| 5 |
C |
0.135 |
|
|
|
| 6 |
C |
0.135 |
|
|
|
| 7 |
H |
0.022 |
|
|
|
| 8 |
H |
0.021 |
|
|
|
| 9 |
H |
0.022 |
|
|
|
| 10 |
H |
0.021 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.360 |
4.360 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.277 |
0.134 |
0.000 |
| y |
0.134 |
-35.928 |
0.000 |
| z |
0.000 |
0.000 |
-35.256 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.316 |
0.134 |
0.000 |
| y |
0.134 |
-2.162 |
0.000 |
| z |
0.000 |
0.000 |
-1.154 |
|
| Polar |
|---|
| 3z2-r2 | -2.308 |
|---|
| x2-y2 | 3.652 |
|---|
| xy | 0.134 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.466 |
0.170 |
0.000 |
| y |
0.170 |
5.393 |
0.000 |
| z |
0.000 |
0.000 |
7.767 |
<r2> (average value of r
2) Å
2
| <r2> |
130.817 |
| (<r2>)1/2 |
11.438 |