Vibrational Frequencies calculated at HF/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3245 |
2948 |
46.65 |
95.09 |
0.42 |
0.59 |
| 2 |
A' |
3216 |
2922 |
54.87 |
83.70 |
0.50 |
0.67 |
| 3 |
A' |
3166 |
2876 |
23.41 |
121.16 |
0.09 |
0.17 |
| 4 |
A' |
3151 |
2863 |
44.93 |
158.91 |
0.00 |
0.01 |
| 5 |
A' |
3124 |
2838 |
88.04 |
179.98 |
0.02 |
0.04 |
| 6 |
A' |
3099 |
2816 |
35.98 |
25.57 |
0.08 |
0.15 |
| 7 |
A' |
1652 |
1501 |
3.76 |
2.94 |
0.73 |
0.84 |
| 8 |
A' |
1627 |
1479 |
5.90 |
2.65 |
0.65 |
0.79 |
| 9 |
A' |
1625 |
1476 |
6.47 |
2.26 |
0.71 |
0.83 |
| 10 |
A' |
1612 |
1464 |
0.27 |
0.78 |
0.57 |
0.73 |
| 11 |
A' |
1611 |
1464 |
1.19 |
9.89 |
0.70 |
0.83 |
| 12 |
A' |
1563 |
1420 |
23.75 |
0.19 |
0.24 |
0.39 |
| 13 |
A' |
1538 |
1397 |
2.20 |
0.28 |
0.25 |
0.39 |
| 14 |
A' |
1455 |
1322 |
5.61 |
1.07 |
0.50 |
0.67 |
| 15 |
A' |
1355 |
1232 |
81.49 |
0.84 |
0.32 |
0.49 |
| 16 |
A' |
1282 |
1165 |
175.08 |
7.80 |
0.34 |
0.51 |
| 17 |
A' |
1215 |
1104 |
10.22 |
4.07 |
0.15 |
0.27 |
| 18 |
A' |
1122 |
1020 |
8.28 |
8.65 |
0.48 |
0.65 |
| 19 |
A' |
1063 |
966 |
23.75 |
2.16 |
0.73 |
0.84 |
| 20 |
A' |
969 |
881 |
3.08 |
7.68 |
0.21 |
0.34 |
| 21 |
A' |
466 |
423 |
0.72 |
4.02 |
0.14 |
0.25 |
| 22 |
A' |
436 |
396 |
3.27 |
1.11 |
0.32 |
0.48 |
| 23 |
A' |
207 |
188 |
1.41 |
0.16 |
0.22 |
0.37 |
| 24 |
A" |
3215 |
2921 |
105.48 |
3.00 |
0.75 |
0.86 |
| 25 |
A" |
3186 |
2895 |
0.02 |
92.79 |
0.75 |
0.86 |
| 26 |
A" |
3161 |
2872 |
77.30 |
55.89 |
0.75 |
0.86 |
| 27 |
A" |
3121 |
2836 |
45.15 |
61.33 |
0.75 |
0.86 |
| 28 |
A" |
1616 |
1468 |
0.82 |
9.88 |
0.75 |
0.86 |
| 29 |
A" |
1616 |
1468 |
11.60 |
0.41 |
0.75 |
0.86 |
| 30 |
A" |
1428 |
1298 |
0.72 |
7.01 |
0.75 |
0.86 |
| 31 |
A" |
1381 |
1254 |
2.50 |
0.04 |
0.75 |
0.86 |
| 32 |
A" |
1303 |
1184 |
8.30 |
0.04 |
0.75 |
0.86 |
| 33 |
A" |
1275 |
1158 |
0.31 |
1.17 |
0.75 |
0.86 |
| 34 |
A" |
968 |
879 |
1.36 |
0.13 |
0.75 |
0.86 |
| 35 |
A" |
816 |
741 |
0.72 |
0.05 |
0.75 |
0.86 |
| 36 |
A" |
253 |
230 |
1.03 |
0.02 |
0.75 |
0.86 |
| 37 |
A" |
235 |
213 |
3.62 |
0.01 |
0.75 |
0.86 |
| 38 |
A" |
123 |
112 |
1.81 |
0.03 |
0.75 |
0.86 |
| 39 |
A" |
106 |
97 |
1.69 |
0.11 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 31799.9 cm
-1
Scaled (by 0.9086) Zero Point Vibrational Energy (zpe) 28893.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
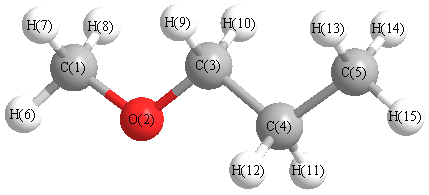
Charges from optimized geometry at HF/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.072 |
|
|
|
| 2 |
O |
-0.812 |
|
|
|
| 3 |
C |
0.074 |
|
|
|
| 4 |
C |
-0.043 |
|
|
|
| 5 |
C |
-0.435 |
|
|
|
| 6 |
H |
0.110 |
|
|
|
| 7 |
H |
0.098 |
|
|
|
| 8 |
H |
0.098 |
|
|
|
| 9 |
H |
0.118 |
|
|
|
| 10 |
H |
0.118 |
|
|
|
| 11 |
H |
0.109 |
|
|
|
| 12 |
H |
0.109 |
|
|
|
| 13 |
H |
0.132 |
|
|
|
| 14 |
H |
0.132 |
|
|
|
| 15 |
H |
0.121 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.247 |
1.173 |
0.000 |
1.199 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.683 |
-2.247 |
0.000 |
| y |
-2.247 |
-33.728 |
0.000 |
| z |
0.000 |
0.000 |
-33.021 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.691 |
-2.247 |
0.000 |
| y |
-2.247 |
-1.376 |
0.000 |
| z |
0.000 |
0.000 |
-0.315 |
|
| Polar |
|---|
| 3z2-r2 | -0.629 |
|---|
| x2-y2 | 2.045 |
|---|
| xy | -2.247 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.238 |
-0.360 |
0.000 |
| y |
-0.360 |
7.307 |
0.000 |
| z |
0.000 |
0.000 |
6.954 |
<r2> (average value of r
2) Å
2
| <r2> |
179.720 |
| (<r2>)1/2 |
13.406 |