Jump to
S1C2
Energy calculated at HF/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -116.933847 |
| Energy at 298.15K | |
| HF Energy | -116.933847 |
| Nuclear repulsion energy | 182.175095 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/CEP-121G
Geometric Data calculated at HF/CEP-121G
Point Group is D4h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
1.017 |
0.000 |
| C2 |
1.017 |
0.000 |
0.000 |
| C3 |
0.000 |
-1.017 |
0.000 |
| C4 |
-1.017 |
0.000 |
0.000 |
| F5 |
0.000 |
2.354 |
0.000 |
| F6 |
2.354 |
0.000 |
0.000 |
| F7 |
0.000 |
-2.354 |
0.000 |
| F8 |
-2.354 |
0.000 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.4384 | 2.0342 | 1.4384 | 1.3369 | 2.5644 | 3.3712 | 2.5644 |
C2 | 1.4384 | | 1.4384 | 2.0342 | 2.5644 | 1.3369 | 2.5644 | 3.3712 | C3 | 2.0342 | 1.4384 | | 1.4384 | 3.3712 | 2.5644 | 1.3369 | 2.5644 | C4 | 1.4384 | 2.0342 | 1.4384 | | 2.5644 | 3.3712 | 2.5644 | 1.3369 | F5 | 1.3369 | 2.5644 | 3.3712 | 2.5644 | | 3.3291 | 4.7081 | 3.3291 | F6 | 2.5644 | 1.3369 | 2.5644 | 3.3712 | 3.3291 | | 3.3291 | 4.7081 | F7 | 3.3712 | 2.5644 | 1.3369 | 2.5644 | 4.7081 | 3.3291 | | 3.3291 | F8 | 2.5644 | 3.3712 | 2.5644 | 1.3369 | 3.3291 | 4.7081 | 3.3291 | |
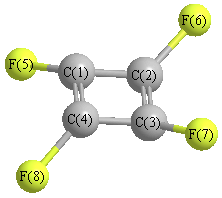 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
135.000 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
135.000 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
135.000 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
135.000 |
| C3 |
C2 |
F6 |
135.000 |
|
C3 |
C4 |
F8 |
135.000 |
| C4 |
C1 |
F5 |
135.000 |
|
C4 |
C3 |
F7 |
135.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/CEP-121G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.592 |
|
|
|
| 2 |
C |
-0.171 |
|
|
|
| 3 |
C |
0.592 |
|
|
|
| 4 |
C |
-0.171 |
|
|
|
| 5 |
F |
-0.184 |
|
|
|
| 6 |
F |
-0.236 |
|
|
|
| 7 |
F |
-0.184 |
|
|
|
| 8 |
F |
-0.236 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-54.833 |
0.000 |
0.000 |
| y |
0.000 |
-43.134 |
0.000 |
| z |
0.000 |
0.000 |
-41.398 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-12.567 |
0.000 |
0.000 |
| y |
0.000 |
4.981 |
0.000 |
| z |
0.000 |
0.000 |
7.585 |
|
| Polar |
|---|
| 3z2-r2 | 15.171 |
|---|
| x2-y2 | -11.699 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.408 |
0.000 |
0.000 |
| y |
0.000 |
7.095 |
0.000 |
| z |
0.000 |
0.000 |
3.414 |
<r2> (average value of r
2) Å
2
| <r2> |
200.730 |
| (<r2>)1/2 |
14.168 |
Jump to
S1C1
Energy calculated at HF/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -117.011236 |
| Energy at 298.15K | -117.011366 |
| HF Energy | -117.011236 |
| Nuclear repulsion energy | 181.763493 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2034 |
1856 |
0.00 |
|
|
|
| 2 |
Ag |
1367 |
1247 |
0.00 |
|
|
|
| 3 |
Ag |
647 |
591 |
0.00 |
|
|
|
| 4 |
Ag |
233 |
212 |
0.00 |
|
|
|
| 5 |
Ag |
154 |
141 |
0.00 |
|
|
|
| 6 |
Au |
1410 |
1287 |
363.83 |
|
|
|
| 7 |
Au |
979 |
893 |
96.85 |
|
|
|
| 8 |
Au |
693 |
632 |
0.00 |
|
|
|
| 9 |
Au |
234 |
213 |
1.45 |
|
|
|
| 10 |
Au |
168 |
153 |
0.00 |
|
|
|
| 11 |
Bg |
1453 |
1326 |
0.00 |
|
|
|
| 12 |
Bg |
782 |
714 |
0.00 |
|
|
|
| 13 |
Bg |
545 |
497 |
0.00 |
|
|
|
| 14 |
Bg |
524 |
478 |
0.00 |
|
|
|
| 15 |
Bu |
2023 |
1846 |
49.80 |
|
|
|
| 16 |
Bu |
992 |
905 |
204.30 |
|
|
|
| 17 |
Bu |
307 |
280 |
16.96 |
|
|
|
| 18 |
Bu |
218 |
199 |
3.80 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 7381.8 cm
-1
Scaled (by 0.9125) Zero Point Vibrational Energy (zpe) 6735.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/CEP-121G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.000 |
0.791 |
0.658 |
| C2 |
0.000 |
-0.791 |
0.658 |
| C3 |
0.000 |
-0.791 |
-0.658 |
| C4 |
-0.000 |
0.791 |
-0.658 |
| F5 |
0.000 |
1.674 |
1.664 |
| F6 |
-0.000 |
-1.674 |
1.664 |
| F7 |
-0.000 |
-1.674 |
-1.664 |
| F8 |
0.000 |
1.674 |
-1.664 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.5821 | 2.0581 | 1.3164 | 1.3383 | 2.6622 | 3.3865 | 2.4843 |
C2 | 1.5821 | | 1.3164 | 2.0581 | 2.6622 | 1.3383 | 2.4843 | 3.3865 | C3 | 2.0581 | 1.3164 | | 1.5821 | 3.3865 | 2.4843 | 1.3383 | 2.6622 | C4 | 1.3164 | 2.0581 | 1.5821 | | 2.4843 | 3.3865 | 2.6622 | 1.3383 | F5 | 1.3383 | 2.6622 | 3.3865 | 2.4843 | | 3.3477 | 4.7204 | 3.3280 | F6 | 2.6622 | 1.3383 | 2.4843 | 3.3865 | 3.3477 | | 3.3280 | 4.7204 | F7 | 3.3865 | 2.4843 | 1.3383 | 2.6622 | 4.7204 | 3.3280 | | 3.3477 | F8 | 2.4843 | 3.3865 | 2.6622 | 1.3383 | 3.3280 | 4.7204 | 3.3477 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
131.275 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
138.725 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
131.275 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
138.725 |
| C3 |
C2 |
F6 |
138.725 |
|
C3 |
C4 |
F8 |
131.275 |
| C4 |
C1 |
F5 |
138.725 |
|
C4 |
C3 |
F7 |
131.275 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/CEP-121G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.223 |
|
|
|
| 2 |
C |
0.223 |
|
|
|
| 3 |
C |
0.223 |
|
|
|
| 4 |
C |
0.223 |
|
|
|
| 5 |
F |
-0.223 |
|
|
|
| 6 |
F |
-0.223 |
|
|
|
| 7 |
F |
-0.223 |
|
|
|
| 8 |
F |
-0.223 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.659 |
-0.001 |
0.000 |
| y |
-0.001 |
-48.855 |
0.000 |
| z |
0.000 |
0.000 |
-49.269 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
8.403 |
-0.001 |
0.000 |
| y |
-0.001 |
-3.891 |
0.000 |
| z |
0.000 |
0.000 |
-4.512 |
|
| Polar |
|---|
| 3z2-r2 | -9.023 |
|---|
| x2-y2 | 8.196 |
|---|
| xy | -0.001 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.164 |
0.000 |
0.000 |
| y |
0.000 |
4.969 |
0.000 |
| z |
0.000 |
0.000 |
7.392 |
<r2> (average value of r
2) Å
2
| <r2> |
201.813 |
| (<r2>)1/2 |
14.206 |