Jump to
S1C2
Vibrational Frequencies calculated at HF/CEP-121G*
Geometric Data calculated at HF/CEP-121G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at HF/CEP-121G*
| | hartrees |
|---|
| Energy at 0K | -39.902384 |
| Energy at 298.15K | -39.916109 |
| HF Energy | -39.902384 |
| Nuclear repulsion energy | 139.542620 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/CEP-121G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3247 |
2934 |
247.77 |
|
|
|
| 2 |
A' |
3235 |
2922 |
39.83 |
|
|
|
| 3 |
A' |
3228 |
2916 |
43.78 |
|
|
|
| 4 |
A' |
3209 |
2899 |
14.03 |
|
|
|
| 5 |
A' |
3191 |
2883 |
11.99 |
|
|
|
| 6 |
A' |
3185 |
2877 |
15.80 |
|
|
|
| 7 |
A' |
3166 |
2860 |
53.33 |
|
|
|
| 8 |
A' |
3163 |
2858 |
38.58 |
|
|
|
| 9 |
A' |
1638 |
1480 |
2.89 |
|
|
|
| 10 |
A' |
1627 |
1470 |
2.87 |
|
|
|
| 11 |
A' |
1624 |
1467 |
4.60 |
|
|
|
| 12 |
A' |
1549 |
1400 |
4.30 |
|
|
|
| 13 |
A' |
1548 |
1399 |
0.97 |
|
|
|
| 14 |
A' |
1503 |
1357 |
4.23 |
|
|
|
| 15 |
A' |
1489 |
1346 |
5.68 |
|
|
|
| 16 |
A' |
1406 |
1270 |
2.03 |
|
|
|
| 17 |
A' |
1352 |
1221 |
0.68 |
|
|
|
| 18 |
A' |
1294 |
1169 |
0.42 |
|
|
|
| 19 |
A' |
1196 |
1080 |
0.44 |
|
|
|
| 20 |
A' |
1114 |
1006 |
0.28 |
|
|
|
| 21 |
A' |
1029 |
930 |
0.35 |
|
|
|
| 22 |
A' |
1014 |
916 |
0.27 |
|
|
|
| 23 |
A' |
921 |
832 |
0.36 |
|
|
|
| 24 |
A' |
847 |
765 |
0.80 |
|
|
|
| 25 |
A' |
692 |
625 |
0.97 |
|
|
|
| 26 |
A' |
457 |
413 |
0.03 |
|
|
|
| 27 |
A' |
341 |
308 |
0.05 |
|
|
|
| 28 |
A' |
101 |
91 |
0.01 |
|
|
|
| 29 |
A" |
3235 |
2923 |
63.44 |
|
|
|
| 30 |
A" |
3230 |
2918 |
33.75 |
|
|
|
| 31 |
A" |
3228 |
2916 |
53.96 |
|
|
|
| 32 |
A" |
3184 |
2876 |
116.70 |
|
|
|
| 33 |
A" |
1628 |
1471 |
2.21 |
|
|
|
| 34 |
A" |
1625 |
1468 |
0.21 |
|
|
|
| 35 |
A" |
1608 |
1453 |
2.39 |
|
|
|
| 36 |
A" |
1422 |
1285 |
1.05 |
|
|
|
| 37 |
A" |
1388 |
1254 |
0.06 |
|
|
|
| 38 |
A" |
1355 |
1224 |
0.07 |
|
|
|
| 39 |
A" |
1240 |
1120 |
0.26 |
|
|
|
| 40 |
A" |
1222 |
1104 |
0.00 |
|
|
|
| 41 |
A" |
1070 |
967 |
1.32 |
|
|
|
| 42 |
A" |
973 |
879 |
0.10 |
|
|
|
| 43 |
A" |
967 |
873 |
0.95 |
|
|
|
| 44 |
A" |
867 |
783 |
0.00 |
|
|
|
| 45 |
A" |
396 |
358 |
0.00 |
|
|
|
| 46 |
A" |
275 |
248 |
0.02 |
|
|
|
| 47 |
A" |
262 |
237 |
0.00 |
|
|
|
| 48 |
A" |
243 |
219 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 38892.0 cm
-1
Scaled (by 0.9034) Zero Point Vibrational Energy (zpe) 35135.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/CEP-121G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.250 |
0.081 |
1.084 |
| C2 |
0.250 |
0.081 |
-1.084 |
| C3 |
-0.661 |
0.724 |
0.000 |
| C4 |
0.845 |
-0.864 |
0.000 |
| C5 |
-0.840 |
2.241 |
0.000 |
| C6 |
0.250 |
-2.276 |
0.000 |
| H7 |
0.988 |
0.787 |
1.460 |
| H8 |
-0.239 |
-0.388 |
1.935 |
| H9 |
0.988 |
0.787 |
-1.460 |
| H10 |
-0.239 |
-0.388 |
-1.935 |
| H11 |
-1.638 |
0.246 |
0.000 |
| H12 |
1.930 |
-0.934 |
0.000 |
| H13 |
0.122 |
2.749 |
0.000 |
| H14 |
-0.838 |
-2.252 |
0.000 |
| H15 |
-1.391 |
2.571 |
-0.880 |
| H16 |
-1.391 |
2.571 |
0.880 |
| H17 |
0.569 |
-2.833 |
-0.880 |
| H18 |
0.569 |
-2.833 |
0.880 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 2.1688 | 1.5552 | 1.5567 | 2.6518 | 2.5948 | 1.0881 | 1.0879 | 2.7417 | 3.0950 | 2.1839 | 2.2422 | 2.8830 | 2.7937 | 3.5711 | 2.9896 | 3.5283 | 2.9380 |
C2 | 2.1688 | | 1.5552 | 1.5567 | 2.6518 | 2.5948 | 2.7417 | 3.0950 | 1.0881 | 1.0879 | 2.1839 | 2.2422 | 2.8830 | 2.7937 | 2.9896 | 3.5711 | 2.9380 | 3.5283 | C3 | 1.5552 | 1.5552 | | 2.1883 | 1.5281 | 3.1352 | 2.2033 | 2.2712 | 2.2033 | 2.2712 | 1.0880 | 3.0756 | 2.1717 | 2.9813 | 2.1728 | 2.1728 | 3.8641 | 3.8641 | C4 | 1.5567 | 1.5567 | 2.1883 | | 3.5335 | 1.5320 | 2.2089 | 2.2691 | 2.2089 | 2.2691 | 2.7200 | 1.0874 | 3.6852 | 2.1815 | 4.1926 | 4.1926 | 2.1735 | 2.1735 | C5 | 2.6518 | 2.6518 | 1.5281 | 3.5335 | | 4.6473 | 2.7552 | 3.3193 | 2.7552 | 3.3193 | 2.1487 | 4.2139 | 1.0887 | 4.4934 | 1.0889 | 1.0889 | 5.3387 | 5.3387 | C6 | 2.5948 | 2.5948 | 3.1352 | 1.5320 | 4.6473 | | 3.4727 | 2.7483 | 3.4727 | 2.7483 | 3.1509 | 2.1504 | 5.0272 | 1.0887 | 5.1931 | 5.1931 | 1.0886 | 1.0886 | H7 | 1.0881 | 2.7417 | 2.2033 | 2.2089 | 2.7552 | 3.4727 | | 1.7641 | 2.9200 | 3.7968 | 3.0533 | 2.4453 | 2.5947 | 3.8346 | 3.7841 | 3.0302 | 4.3304 | 3.6895 | H8 | 1.0879 | 3.0950 | 2.2712 | 2.2691 | 3.3193 | 2.7483 | 1.7641 | | 3.7968 | 3.8710 | 2.4707 | 2.9581 | 3.7036 | 2.7536 | 4.2434 | 3.3462 | 3.8154 | 2.7828 | H9 | 2.7417 | 1.0881 | 2.2033 | 2.2089 | 2.7552 | 3.4727 | 2.9200 | 3.7968 | | 1.7641 | 3.0533 | 2.4453 | 2.5947 | 3.8346 | 3.0302 | 3.7841 | 3.6895 | 4.3304 | H10 | 3.0950 | 1.0879 | 2.2712 | 2.2691 | 3.3193 | 2.7483 | 3.7968 | 3.8710 | 1.7641 | | 2.4707 | 2.9581 | 3.7036 | 2.7536 | 3.3462 | 4.2434 | 2.7828 | 3.8154 | H11 | 2.1839 | 2.1839 | 1.0880 | 2.7200 | 2.1487 | 3.1509 | 3.0533 | 2.4707 | 3.0533 | 2.4707 | | 3.7583 | 3.0604 | 2.6232 | 2.4985 | 2.4985 | 3.8886 | 3.8886 | H12 | 2.2422 | 2.2422 | 3.0756 | 1.0874 | 4.2139 | 2.1504 | 2.4453 | 2.9581 | 2.4453 | 2.9581 | 3.7583 | | 4.1028 | 3.0662 | 4.9081 | 4.9081 | 2.4967 | 2.4967 | H13 | 2.8830 | 2.8830 | 2.1717 | 3.6852 | 1.0887 | 5.0272 | 2.5947 | 3.7036 | 2.5947 | 3.7036 | 3.0604 | 4.1028 | | 5.0929 | 1.7595 | 1.7595 | 5.6683 | 5.6683 | H14 | 2.7937 | 2.7937 | 2.9813 | 2.1815 | 4.4934 | 1.0887 | 3.8346 | 2.7536 | 3.8346 | 2.7536 | 2.6232 | 3.0662 | 5.0929 | | 4.9343 | 4.9343 | 1.7578 | 1.7578 | H15 | 3.5711 | 2.9896 | 2.1728 | 4.1926 | 1.0889 | 5.1931 | 3.7841 | 4.2434 | 3.0302 | 3.3462 | 2.4985 | 4.9081 | 1.7595 | 4.9343 | | 1.7592 | 5.7482 | 6.0115 | H16 | 2.9896 | 3.5711 | 2.1728 | 4.1926 | 1.0889 | 5.1931 | 3.0302 | 3.3462 | 3.7841 | 4.2434 | 2.4985 | 4.9081 | 1.7595 | 4.9343 | 1.7592 | | 6.0115 | 5.7482 | H17 | 3.5283 | 2.9380 | 3.8641 | 2.1735 | 5.3387 | 1.0886 | 4.3304 | 3.8154 | 3.6895 | 2.7828 | 3.8886 | 2.4967 | 5.6683 | 1.7578 | 5.7482 | 6.0115 | | 1.7601 | H18 | 2.9380 | 3.5283 | 3.8641 | 2.1735 | 5.3387 | 1.0886 | 3.6895 | 2.7828 | 4.3304 | 3.8154 | 3.8886 | 2.4967 | 5.6683 | 1.7578 | 6.0115 | 5.7482 | 1.7601 | |
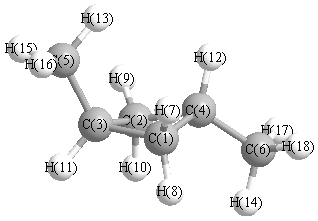 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
C2 |
88.418 |
|
C1 |
C3 |
C5 |
118.640 |
| C1 |
C3 |
H11 |
110.170 |
|
C1 |
C4 |
C2 |
88.309 |
| C1 |
C4 |
C6 |
114.297 |
|
C1 |
C4 |
H12 |
114.829 |
| C2 |
C3 |
C5 |
118.640 |
|
C2 |
C3 |
H11 |
110.170 |
| C2 |
C4 |
C6 |
114.297 |
|
C2 |
C4 |
H12 |
114.829 |
| C3 |
C1 |
C4 |
89.368 |
|
C3 |
C1 |
H7 |
111.707 |
| C3 |
C1 |
H8 |
117.383 |
|
C3 |
C2 |
C4 |
89.368 |
| C3 |
C2 |
H9 |
111.707 |
|
C3 |
C2 |
H10 |
117.383 |
| C3 |
C5 |
H13 |
111.058 |
|
C3 |
C5 |
H15 |
111.142 |
| C3 |
C5 |
H16 |
111.142 |
|
C4 |
C1 |
H7 |
112.046 |
| C4 |
C1 |
H8 |
117.081 |
|
C4 |
C2 |
H9 |
112.046 |
| C4 |
C2 |
H10 |
117.081 |
|
C4 |
C6 |
H14 |
111.574 |
| C4 |
C6 |
H17 |
110.940 |
|
C4 |
C6 |
H18 |
110.940 |
| C5 |
C3 |
H11 |
109.282 |
|
C6 |
C4 |
H12 |
109.183 |
| H7 |
C1 |
H8 |
108.334 |
|
H9 |
C2 |
H10 |
108.334 |
| H13 |
C5 |
H15 |
107.792 |
|
H13 |
C5 |
H16 |
107.792 |
| H14 |
C6 |
H17 |
107.668 |
|
H14 |
C6 |
H18 |
107.668 |
| H15 |
C5 |
H16 |
107.755 |
|
H17 |
C6 |
H18 |
107.883 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/CEP-121G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.471 |
|
|
|
| 2 |
C |
-0.471 |
|
|
|
| 3 |
C |
0.055 |
|
|
|
| 4 |
C |
0.093 |
|
|
|
| 5 |
C |
-0.581 |
|
|
|
| 6 |
C |
-0.596 |
|
|
|
| 7 |
H |
0.166 |
|
|
|
| 8 |
H |
0.167 |
|
|
|
| 9 |
H |
0.166 |
|
|
|
| 10 |
H |
0.167 |
|
|
|
| 11 |
H |
0.172 |
|
|
|
| 12 |
H |
0.177 |
|
|
|
| 13 |
H |
0.154 |
|
|
|
| 14 |
H |
0.159 |
|
|
|
| 15 |
H |
0.159 |
|
|
|
| 16 |
H |
0.159 |
|
|
|
| 17 |
H |
0.162 |
|
|
|
| 18 |
H |
0.162 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.002 |
-0.015 |
0.000 |
0.015 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-39.701 |
0.361 |
0.000 |
| y |
0.361 |
-41.229 |
0.000 |
| z |
0.000 |
0.000 |
-40.627 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.227 |
0.361 |
0.000 |
| y |
0.361 |
-1.065 |
0.000 |
| z |
0.000 |
0.000 |
-0.162 |
|
| Polar |
|---|
| 3z2-r2 | -0.323 |
|---|
| x2-y2 | 1.528 |
|---|
| xy | 0.361 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.903 |
-0.601 |
0.000 |
| y |
-0.601 |
10.424 |
0.000 |
| z |
0.000 |
0.000 |
9.329 |
<r2> (average value of r
2) Å
2
| <r2> |
161.783 |
| (<r2>)1/2 |
12.719 |