Jump to
S1C2
Energy calculated at HF/CEP-121G*
| | hartrees |
|---|
| Energy at 0K | -69.529759 |
| Energy at 298.15K | -69.534824 |
| Nuclear repulsion energy | 126.271312 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/CEP-121G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
4079 |
3685 |
116.71 |
|
|
|
| 2 |
A' |
3969 |
3586 |
88.27 |
|
|
|
| 3 |
A' |
3826 |
3456 |
79.89 |
|
|
|
| 4 |
A' |
1998 |
1805 |
9.47 |
|
|
|
| 5 |
A' |
1951 |
1763 |
987.41 |
|
|
|
| 6 |
A' |
1773 |
1602 |
119.24 |
|
|
|
| 7 |
A' |
1564 |
1413 |
15.10 |
|
|
|
| 8 |
A' |
1423 |
1286 |
57.89 |
|
|
|
| 9 |
A' |
1319 |
1192 |
428.55 |
|
|
|
| 10 |
A' |
1193 |
1078 |
10.35 |
|
|
|
| 11 |
A' |
849 |
767 |
11.71 |
|
|
|
| 12 |
A' |
661 |
597 |
97.96 |
|
|
|
| 13 |
A' |
570 |
515 |
0.06 |
|
|
|
| 14 |
A' |
444 |
401 |
3.22 |
|
|
|
| 15 |
A' |
289 |
261 |
15.91 |
|
|
|
| 16 |
A" |
915 |
826 |
6.74 |
|
|
|
| 17 |
A" |
700 |
633 |
176.10 |
|
|
|
| 18 |
A" |
645 |
583 |
35.34 |
|
|
|
| 19 |
A" |
472 |
426 |
3.03 |
|
|
|
| 20 |
A" |
251 |
227 |
308.14 |
|
|
|
| 21 |
A" |
20 |
18 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14455.3 cm
-1
Scaled (by 0.9034) Zero Point Vibrational Energy (zpe) 13059.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/CEP-121G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.761 |
0.000 |
| C2 |
-0.057 |
-0.791 |
0.000 |
| O3 |
-1.093 |
-1.387 |
0.000 |
| O4 |
1.030 |
1.365 |
0.000 |
| O5 |
-1.195 |
1.318 |
0.000 |
| N6 |
1.177 |
-1.336 |
0.000 |
| H7 |
1.260 |
-2.329 |
0.000 |
| H8 |
1.991 |
-0.764 |
0.000 |
| H9 |
-1.081 |
2.263 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5528 | 2.4108 | 1.1937 | 1.3182 | 2.4052 | 3.3372 | 2.5080 | 1.8508 |
C2 | 1.5528 | | 1.1960 | 2.4139 | 2.3958 | 1.3490 | 2.0250 | 2.0477 | 3.2211 | O3 | 2.4108 | 1.1960 | | 3.4760 | 2.7070 | 2.2706 | 2.5346 | 3.1463 | 3.6508 | O4 | 1.1937 | 2.4139 | 3.4760 | | 2.2252 | 2.7055 | 3.7012 | 2.3360 | 2.2942 | O5 | 1.3182 | 2.3958 | 2.7070 | 2.2252 | | 3.5594 | 4.3960 | 3.8056 | 0.9525 | N6 | 2.4052 | 1.3490 | 2.2706 | 2.7055 | 3.5594 | | 0.9960 | 0.9950 | 4.2494 | H7 | 3.3372 | 2.0250 | 2.5346 | 3.7012 | 4.3960 | 0.9960 | | 1.7271 | 5.1547 | H8 | 2.5080 | 2.0477 | 3.1463 | 2.3360 | 3.8056 | 0.9950 | 1.7271 | | 4.3131 | H9 | 1.8508 | 3.2211 | 3.6508 | 2.2942 | 0.9525 | 4.2494 | 5.1547 | 4.3131 | |
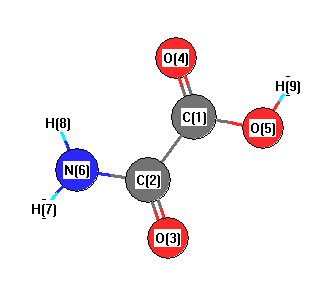 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
122.035 |
|
C1 |
C2 |
N6 |
111.777 |
| C1 |
O5 |
H9 |
108.114 |
|
C2 |
C1 |
O4 |
122.482 |
| C2 |
C1 |
O5 |
112.872 |
|
C2 |
N6 |
H7 |
118.663 |
| C2 |
N6 |
H8 |
121.014 |
|
O3 |
C2 |
N6 |
126.188 |
| O4 |
C1 |
O5 |
124.646 |
|
H7 |
N6 |
H8 |
120.323 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/CEP-121G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.376 |
|
|
|
| 2 |
C |
0.320 |
|
|
|
| 3 |
O |
-0.385 |
|
|
|
| 4 |
O |
-0.417 |
|
|
|
| 5 |
O |
-0.492 |
|
|
|
| 6 |
N |
-0.693 |
|
|
|
| 7 |
H |
0.396 |
|
|
|
| 8 |
H |
0.422 |
|
|
|
| 9 |
H |
0.472 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.313 |
0.736 |
0.000 |
2.427 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.665 |
-7.398 |
0.003 |
| y |
-7.398 |
-41.104 |
-0.001 |
| z |
0.003 |
-0.001 |
-33.599 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
13.686 |
-7.398 |
0.003 |
| y |
-7.398 |
-12.472 |
-0.001 |
| z |
0.003 |
-0.001 |
-1.214 |
|
| Polar |
|---|
| 3z2-r2 | -2.429 |
|---|
| x2-y2 | 17.438 |
|---|
| xy | -7.398 |
|---|
| xz | 0.003 |
|---|
| yz | -0.001 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.408 |
-0.088 |
-0.000 |
| y |
-0.088 |
6.271 |
0.000 |
| z |
-0.000 |
0.000 |
3.097 |
<r2> (average value of r
2) Å
2
| <r2> |
114.264 |
| (<r2>)1/2 |
10.689 |
Jump to
S1C1
Energy calculated at HF/CEP-121G*
| | hartrees |
|---|
| Energy at 0K | -69.534087 |
| Energy at 298.15K | -69.539480 |
| Nuclear repulsion energy | 127.081025 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/CEP-121G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
4023 |
3634 |
164.91 |
|
|
|
| 2 |
A' |
3960 |
3577 |
93.38 |
|
|
|
| 3 |
A' |
3817 |
3448 |
89.25 |
|
|
|
| 4 |
A' |
2029 |
1833 |
252.02 |
|
|
|
| 5 |
A' |
1928 |
1742 |
687.38 |
|
|
|
| 6 |
A' |
1775 |
1604 |
80.97 |
|
|
|
| 7 |
A' |
1586 |
1433 |
173.82 |
|
|
|
| 8 |
A' |
1446 |
1306 |
535.39 |
|
|
|
| 9 |
A' |
1311 |
1184 |
32.73 |
|
|
|
| 10 |
A' |
1197 |
1082 |
0.23 |
|
|
|
| 11 |
A' |
877 |
793 |
9.73 |
|
|
|
| 12 |
A' |
677 |
612 |
26.00 |
|
|
|
| 13 |
A' |
586 |
529 |
3.84 |
|
|
|
| 14 |
A' |
440 |
398 |
6.56 |
|
|
|
| 15 |
A' |
294 |
266 |
39.56 |
|
|
|
| 16 |
A" |
908 |
820 |
0.26 |
|
|
|
| 17 |
A" |
711 |
642 |
159.43 |
|
|
|
| 18 |
A" |
683 |
617 |
8.26 |
|
|
|
| 19 |
A" |
501 |
453 |
100.16 |
|
|
|
| 20 |
A" |
342 |
309 |
253.45 |
|
|
|
| 21 |
A" |
95 |
85 |
3.76 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14592.4 cm
-1
Scaled (by 0.9034) Zero Point Vibrational Energy (zpe) 13182.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/CEP-121G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.003 |
-0.799 |
0.000 |
| C2 |
0.000 |
0.754 |
0.000 |
| O3 |
-1.054 |
1.340 |
0.000 |
| O4 |
1.007 |
-1.433 |
0.000 |
| O5 |
-1.210 |
-1.312 |
0.000 |
| N6 |
1.222 |
1.300 |
0.000 |
| H7 |
1.315 |
2.292 |
0.000 |
| H8 |
2.030 |
0.718 |
0.000 |
| H9 |
-1.848 |
-0.601 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5527 | 2.3857 | 1.1877 | 1.3172 | 2.4267 | 3.3575 | 2.5316 | 1.8617 |
C2 | 1.5527 | | 1.2062 | 2.4080 | 2.3941 | 1.3380 | 2.0233 | 2.0303 | 2.2917 | O3 | 2.3857 | 1.2062 | | 3.4554 | 2.6562 | 2.2763 | 2.5532 | 3.1465 | 2.0970 | O4 | 1.1877 | 2.4080 | 3.4554 | | 2.2205 | 2.7416 | 3.7380 | 2.3821 | 2.9740 | O5 | 1.3172 | 2.3941 | 2.6562 | 2.2205 | | 3.5684 | 4.4001 | 3.8234 | 0.9550 | N6 | 2.4267 | 1.3380 | 2.2763 | 2.7416 | 3.5684 | | 0.9965 | 0.9961 | 3.6106 | H7 | 3.3575 | 2.0233 | 2.5532 | 3.7380 | 4.4001 | 0.9965 | | 1.7290 | 4.2864 | H8 | 2.5316 | 2.0303 | 3.1465 | 2.3821 | 3.8234 | 0.9961 | 1.7290 | | 4.0963 | H9 | 1.8617 | 2.2917 | 2.0970 | 2.9740 | 0.9550 | 3.6106 | 4.2864 | 4.0963 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.169 |
|
C1 |
C2 |
N6 |
113.965 |
| C1 |
O5 |
H9 |
108.996 |
|
C2 |
C1 |
O4 |
122.408 |
| C2 |
C1 |
O5 |
112.810 |
|
C2 |
N6 |
H7 |
119.438 |
| C2 |
N6 |
H8 |
120.177 |
|
O3 |
C2 |
N6 |
126.866 |
| O4 |
C1 |
O5 |
124.782 |
|
H7 |
N6 |
H8 |
120.385 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/CEP-121G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.391 |
|
|
|
| 2 |
C |
0.304 |
|
|
|
| 3 |
O |
-0.458 |
|
|
|
| 4 |
O |
-0.384 |
|
|
|
| 5 |
O |
-0.524 |
|
|
|
| 6 |
N |
-0.662 |
|
|
|
| 7 |
H |
0.399 |
|
|
|
| 8 |
H |
0.428 |
|
|
|
| 9 |
H |
0.506 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.419 |
2.616 |
0.000 |
2.976 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.484 |
7.859 |
0.000 |
| y |
7.859 |
-38.785 |
0.000 |
| z |
0.000 |
0.000 |
-33.555 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.686 |
7.859 |
0.000 |
| y |
7.859 |
-6.766 |
0.000 |
| z |
0.000 |
0.000 |
1.080 |
|
| Polar |
|---|
| 3z2-r2 | 2.160 |
|---|
| x2-y2 | 8.302 |
|---|
| xy | 7.859 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.475 |
-0.265 |
0.000 |
| y |
-0.265 |
5.091 |
0.000 |
| z |
0.000 |
0.000 |
3.074 |
<r2> (average value of r
2) Å
2
| <r2> |
112.501 |
| (<r2>)1/2 |
10.607 |