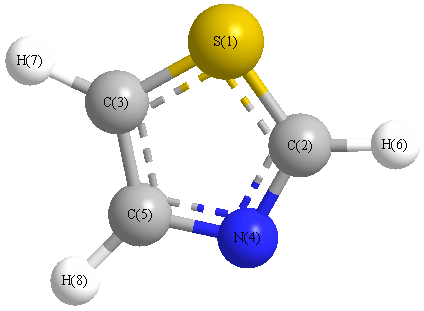
Vibrational Frequencies calculated at HF/LANL2DZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3509 |
3158 |
4.41 |
137.80 |
0.17 |
0.29 |
| 2 |
A' |
3488 |
3139 |
0.08 |
78.82 |
0.26 |
0.41 |
| 3 |
A' |
3476 |
3128 |
4.63 |
74.80 |
0.63 |
0.77 |
| 4 |
A' |
1683 |
1515 |
29.29 |
4.24 |
0.43 |
0.60 |
| 5 |
A' |
1609 |
1448 |
45.88 |
52.86 |
0.17 |
0.29 |
| 6 |
A' |
1455 |
1309 |
3.78 |
6.23 |
0.59 |
0.74 |
| 7 |
A' |
1368 |
1231 |
18.28 |
1.78 |
0.45 |
0.62 |
| 8 |
A' |
1245 |
1120 |
6.89 |
10.00 |
0.64 |
0.78 |
| 9 |
A' |
1120 |
1008 |
13.02 |
10.48 |
0.43 |
0.60 |
| 10 |
A' |
959 |
863 |
13.17 |
0.98 |
0.57 |
0.73 |
| 11 |
A' |
878 |
790 |
52.34 |
17.28 |
0.30 |
0.46 |
| 12 |
A' |
750 |
675 |
2.26 |
8.14 |
0.74 |
0.85 |
| 13 |
A' |
650 |
585 |
7.66 |
17.82 |
0.30 |
0.46 |
| 14 |
A" |
1058 |
952 |
0.11 |
2.95 |
0.75 |
0.86 |
| 15 |
A" |
931 |
838 |
43.80 |
1.16 |
0.75 |
0.86 |
| 16 |
A" |
857 |
771 |
63.68 |
0.42 |
0.75 |
0.86 |
| 17 |
A" |
670 |
603 |
32.94 |
5.21 |
0.75 |
0.86 |
| 18 |
A" |
490 |
441 |
1.46 |
0.74 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 13098.1 cm
-1
Scaled (by 0.8999) Zero Point Vibrational Energy (zpe) 11787.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/LANL2DZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.247 |
|
|
|
| 2 |
C |
-0.319 |
|
|
|
| 3 |
C |
-0.481 |
|
|
|
| 4 |
N |
-0.158 |
|
|
|
| 5 |
C |
-0.051 |
|
|
|
| 6 |
H |
0.258 |
|
|
|
| 7 |
H |
0.256 |
|
|
|
| 8 |
H |
0.248 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.167 |
0.843 |
0.000 |
1.440 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.528 |
-4.317 |
0.000 |
| y |
-4.317 |
-40.090 |
0.000 |
| z |
0.000 |
0.000 |
-39.096 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
9.065 |
-4.317 |
0.000 |
| y |
-4.317 |
-5.278 |
0.000 |
| z |
0.000 |
0.000 |
-3.787 |
|
| Polar |
|---|
| 3z2-r2 | -7.575 |
|---|
| x2-y2 | 9.562 |
|---|
| xy | -4.317 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.831 |
-0.057 |
0.000 |
| y |
-0.057 |
8.544 |
0.000 |
| z |
0.000 |
0.000 |
2.565 |
<r2> (average value of r
2) Å
2
| <r2> |
94.438 |
| (<r2>)1/2 |
9.718 |