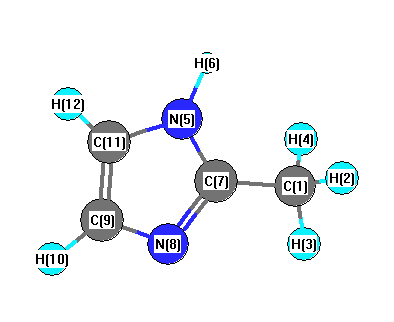
Vibrational Frequencies calculated at HF/LANL2DZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3948 |
3553 |
88.68 |
|
|
|
| 2 |
A |
3518 |
3165 |
7.84 |
|
|
|
| 3 |
A |
3490 |
3141 |
3.74 |
|
|
|
| 4 |
A |
3342 |
3007 |
12.23 |
|
|
|
| 5 |
A |
3276 |
2948 |
38.60 |
|
|
|
| 6 |
A |
3207 |
2886 |
45.81 |
|
|
|
| 7 |
A |
1752 |
1577 |
66.43 |
|
|
|
| 8 |
A |
1658 |
1492 |
5.34 |
|
|
|
| 9 |
A |
1640 |
1476 |
5.55 |
|
|
|
| 10 |
A |
1640 |
1476 |
14.31 |
|
|
|
| 11 |
A |
1577 |
1419 |
1.07 |
|
|
|
| 12 |
A |
1558 |
1402 |
49.94 |
|
|
|
| 13 |
A |
1493 |
1344 |
2.20 |
|
|
|
| 14 |
A |
1398 |
1258 |
24.72 |
|
|
|
| 15 |
A |
1261 |
1135 |
17.34 |
|
|
|
| 16 |
A |
1216 |
1094 |
3.47 |
|
|
|
| 17 |
A |
1198 |
1078 |
3.82 |
|
|
|
| 18 |
A |
1190 |
1071 |
33.25 |
|
|
|
| 19 |
A |
1099 |
989 |
17.75 |
|
|
|
| 20 |
A |
1062 |
956 |
8.14 |
|
|
|
| 21 |
A |
1023 |
920 |
4.97 |
|
|
|
| 22 |
A |
1017 |
915 |
1.83 |
|
|
|
| 23 |
A |
866 |
779 |
78.54 |
|
|
|
| 24 |
A |
763 |
687 |
22.27 |
|
|
|
| 25 |
A |
723 |
650 |
2.32 |
|
|
|
| 26 |
A |
715 |
643 |
152.71 |
|
|
|
| 27 |
A |
694 |
625 |
11.12 |
|
|
|
| 28 |
A |
371 |
334 |
6.86 |
|
|
|
| 29 |
A |
284 |
255 |
9.42 |
|
|
|
| 30 |
A |
93 |
84 |
0.30 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23535.1 cm
-1
Scaled (by 0.8999) Zero Point Vibrational Energy (zpe) 21179.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/LANL2DZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.615 |
|
|
|
| 2 |
H |
0.189 |
|
|
|
| 3 |
H |
0.236 |
|
|
|
| 4 |
H |
0.189 |
|
|
|
| 5 |
N |
-0.602 |
|
|
|
| 6 |
H |
0.378 |
|
|
|
| 7 |
C |
0.366 |
|
|
|
| 8 |
N |
-0.268 |
|
|
|
| 9 |
C |
-0.204 |
|
|
|
| 10 |
H |
0.227 |
|
|
|
| 11 |
C |
-0.127 |
|
|
|
| 12 |
H |
0.232 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.547 |
4.108 |
-0.001 |
4.145 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.308 |
0.760 |
-0.001 |
| y |
0.760 |
-35.756 |
-0.004 |
| z |
-0.001 |
-0.004 |
-39.179 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
7.159 |
0.760 |
-0.001 |
| y |
0.760 |
-1.013 |
-0.004 |
| z |
-0.001 |
-0.004 |
-6.147 |
|
| Polar |
|---|
| 3z2-r2 | -12.293 |
|---|
| x2-y2 | 5.448 |
|---|
| xy | 0.760 |
|---|
| xz | -0.001 |
|---|
| yz | -0.004 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.687 |
0.152 |
0.000 |
| y |
0.152 |
7.810 |
-0.000 |
| z |
0.000 |
-0.000 |
4.097 |
<r2> (average value of r
2) Å
2
| <r2> |
139.976 |
| (<r2>)1/2 |
11.831 |