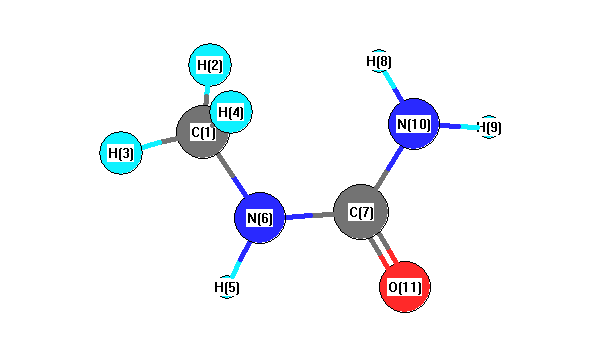
Vibrational Frequencies calculated at HF/SDD
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
4028 |
3626 |
62.10 |
|
|
|
| 2 |
A |
3912 |
3522 |
58.39 |
|
|
|
| 3 |
A |
3875 |
3489 |
62.99 |
|
|
|
| 4 |
A |
3322 |
2991 |
35.18 |
|
|
|
| 5 |
A |
3264 |
2939 |
67.68 |
|
|
|
| 6 |
A |
3199 |
2880 |
60.90 |
|
|
|
| 7 |
A |
1843 |
1660 |
529.42 |
|
|
|
| 8 |
A |
1794 |
1616 |
291.23 |
|
|
|
| 9 |
A |
1676 |
1509 |
35.54 |
|
|
|
| 10 |
A |
1654 |
1489 |
10.76 |
|
|
|
| 11 |
A |
1639 |
1476 |
105.24 |
|
|
|
| 12 |
A |
1594 |
1435 |
45.06 |
|
|
|
| 13 |
A |
1565 |
1410 |
264.42 |
|
|
|
| 14 |
A |
1308 |
1178 |
1.70 |
|
|
|
| 15 |
A |
1264 |
1138 |
0.32 |
|
|
|
| 16 |
A |
1233 |
1110 |
57.72 |
|
|
|
| 17 |
A |
1181 |
1064 |
23.14 |
|
|
|
| 18 |
A |
949 |
855 |
5.92 |
|
|
|
| 19 |
A |
867 |
780 |
226.18 |
|
|
|
| 20 |
A |
622 |
560 |
295.58 |
|
|
|
| 21 |
A |
614 |
553 |
12.63 |
|
|
|
| 22 |
A |
599 |
539 |
11.10 |
|
|
|
| 23 |
A |
567 |
511 |
18.19 |
|
|
|
| 24 |
A |
480 |
432 |
170.74 |
|
|
|
| 25 |
A |
309 |
278 |
5.21 |
|
|
|
| 26 |
A |
176 |
158 |
1.33 |
|
|
|
| 27 |
A |
140 |
126 |
0.94 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21835.7 cm
-1
Scaled (by 0.9004) Zero Point Vibrational Energy (zpe) 19660.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/SDD
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.440 |
|
|
|
| 2 |
H |
0.201 |
|
|
|
| 3 |
H |
0.205 |
|
|
|
| 4 |
H |
0.201 |
|
|
|
| 5 |
H |
0.392 |
|
|
|
| 6 |
N |
-0.598 |
|
|
|
| 7 |
C |
0.601 |
|
|
|
| 8 |
H |
0.379 |
|
|
|
| 9 |
H |
0.396 |
|
|
|
| 10 |
N |
-0.833 |
|
|
|
| 11 |
O |
-0.503 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
4.737 |
2.882 |
-0.002 |
5.544 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.307 |
-6.533 |
0.003 |
| y |
-6.533 |
-26.211 |
-0.001 |
| z |
0.003 |
-0.001 |
-32.577 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.913 |
-6.533 |
0.003 |
| y |
-6.533 |
6.732 |
-0.001 |
| z |
0.003 |
-0.001 |
-2.818 |
|
| Polar |
|---|
| 3z2-r2 | -5.637 |
|---|
| x2-y2 | -7.097 |
|---|
| xy | -6.533 |
|---|
| xz | 0.003 |
|---|
| yz | -0.001 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.065 |
0.316 |
-0.001 |
| y |
0.316 |
5.225 |
-0.000 |
| z |
-0.001 |
-0.000 |
3.260 |
<r2> (average value of r
2) Å
2
| <r2> |
120.387 |
| (<r2>)1/2 |
10.972 |