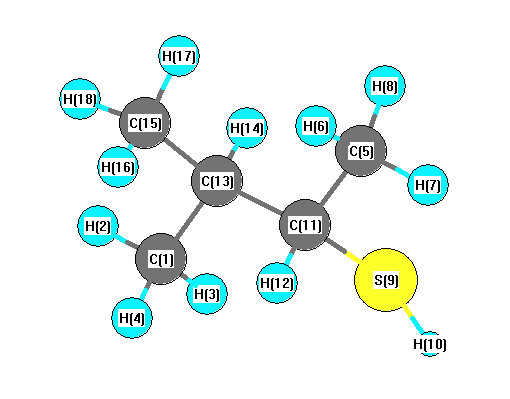
Vibrational Frequencies calculated at HF/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3244 |
2953 |
29.96 |
|
|
|
| 2 |
A |
3241 |
2951 |
46.03 |
|
|
|
| 3 |
A |
3232 |
2942 |
49.61 |
|
|
|
| 4 |
A |
3225 |
2936 |
42.80 |
|
|
|
| 5 |
A |
3217 |
2929 |
80.94 |
|
|
|
| 6 |
A |
3205 |
2917 |
1.42 |
|
|
|
| 7 |
A |
3186 |
2900 |
17.65 |
|
|
|
| 8 |
A |
3167 |
2884 |
21.93 |
|
|
|
| 9 |
A |
3158 |
2875 |
39.50 |
|
|
|
| 10 |
A |
3152 |
2869 |
16.56 |
|
|
|
| 11 |
A |
3144 |
2862 |
3.61 |
|
|
|
| 12 |
A |
2857 |
2601 |
5.07 |
|
|
|
| 13 |
A |
1639 |
1492 |
2.38 |
|
|
|
| 14 |
A |
1623 |
1477 |
7.74 |
|
|
|
| 15 |
A |
1622 |
1476 |
12.89 |
|
|
|
| 16 |
A |
1611 |
1467 |
2.24 |
|
|
|
| 17 |
A |
1610 |
1465 |
3.06 |
|
|
|
| 18 |
A |
1604 |
1460 |
1.15 |
|
|
|
| 19 |
A |
1550 |
1411 |
4.54 |
|
|
|
| 20 |
A |
1540 |
1402 |
5.31 |
|
|
|
| 21 |
A |
1531 |
1393 |
2.36 |
|
|
|
| 22 |
A |
1510 |
1375 |
1.24 |
|
|
|
| 23 |
A |
1470 |
1338 |
0.48 |
|
|
|
| 24 |
A |
1438 |
1309 |
4.02 |
|
|
|
| 25 |
A |
1366 |
1244 |
31.34 |
|
|
|
| 26 |
A |
1294 |
1178 |
2.39 |
|
|
|
| 27 |
A |
1278 |
1164 |
8.44 |
|
|
|
| 28 |
A |
1236 |
1125 |
0.89 |
|
|
|
| 29 |
A |
1167 |
1063 |
2.93 |
|
|
|
| 30 |
A |
1118 |
1017 |
7.75 |
|
|
|
| 31 |
A |
1064 |
969 |
1.19 |
|
|
|
| 32 |
A |
1038 |
945 |
0.10 |
|
|
|
| 33 |
A |
1006 |
915 |
0.50 |
|
|
|
| 34 |
A |
982 |
894 |
1.93 |
|
|
|
| 35 |
A |
968 |
881 |
2.79 |
|
|
|
| 36 |
A |
843 |
767 |
3.09 |
|
|
|
| 37 |
A |
737 |
671 |
4.15 |
|
|
|
| 38 |
A |
508 |
462 |
0.47 |
|
|
|
| 39 |
A |
434 |
395 |
0.51 |
|
|
|
| 40 |
A |
400 |
364 |
0.12 |
|
|
|
| 41 |
A |
385 |
351 |
0.15 |
|
|
|
| 42 |
A |
357 |
325 |
1.19 |
|
|
|
| 43 |
A |
275 |
250 |
1.01 |
|
|
|
| 44 |
A |
254 |
232 |
3.07 |
|
|
|
| 45 |
A |
245 |
223 |
0.32 |
|
|
|
| 46 |
A |
210 |
191 |
1.09 |
|
|
|
| 47 |
A |
185 |
169 |
13.43 |
|
|
|
| 48 |
A |
62 |
56 |
1.82 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 37091.1 cm
-1
Scaled (by 0.9104) Zero Point Vibrational Energy (zpe) 33767.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-1.136 |
|
|
|
| 2 |
H |
0.257 |
|
|
|
| 3 |
H |
0.299 |
|
|
|
| 4 |
H |
0.253 |
|
|
|
| 5 |
C |
-1.057 |
|
|
|
| 6 |
H |
0.266 |
|
|
|
| 7 |
H |
0.295 |
|
|
|
| 8 |
H |
0.300 |
|
|
|
| 9 |
S |
-0.524 |
|
|
|
| 10 |
H |
-0.004 |
|
|
|
| 11 |
C |
0.293 |
|
|
|
| 12 |
H |
0.292 |
|
|
|
| 13 |
C |
0.357 |
|
|
|
| 14 |
H |
0.380 |
|
|
|
| 15 |
C |
-1.073 |
|
|
|
| 16 |
H |
0.271 |
|
|
|
| 17 |
H |
0.278 |
|
|
|
| 18 |
H |
0.252 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.034 |
1.366 |
0.637 |
1.827 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.858 |
2.640 |
1.906 |
| y |
2.640 |
-49.015 |
0.804 |
| z |
1.906 |
0.804 |
-48.458 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.878 |
2.640 |
1.906 |
| y |
2.640 |
-0.857 |
0.804 |
| z |
1.906 |
0.804 |
-0.021 |
|
| Polar |
|---|
| 3z2-r2 | -0.043 |
|---|
| x2-y2 | 1.157 |
|---|
| xy | 2.640 |
|---|
| xz | 1.906 |
|---|
| yz | 0.804 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.366 |
-0.347 |
-0.066 |
| y |
-0.347 |
12.103 |
-0.005 |
| z |
-0.066 |
-0.005 |
10.115 |
<r2> (average value of r
2) Å
2
| <r2> |
250.164 |
| (<r2>)1/2 |
15.817 |