Jump to
S1C2
Energy calculated at HF/3-21G
| | hartrees |
|---|
| Energy at 0K | -266.474941 |
| Energy at 298.15K | -266.484742 |
| Nuclear repulsion energy | 197.171557 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3307 |
2995 |
1.96 |
|
|
|
| 2 |
A |
3263 |
2955 |
69.04 |
|
|
|
| 3 |
A |
3224 |
2919 |
75.48 |
|
|
|
| 4 |
A |
3190 |
2889 |
1.07 |
|
|
|
| 5 |
A |
1701 |
1541 |
3.56 |
|
|
|
| 6 |
A |
1689 |
1530 |
0.36 |
|
|
|
| 7 |
A |
1669 |
1511 |
6.82 |
|
|
|
| 8 |
A |
1614 |
1462 |
1.54 |
|
|
|
| 9 |
A |
1451 |
1314 |
0.93 |
|
|
|
| 10 |
A |
1299 |
1176 |
33.95 |
|
|
|
| 11 |
A |
1271 |
1151 |
10.86 |
|
|
|
| 12 |
A |
1211 |
1097 |
110.93 |
|
|
|
| 13 |
A |
969 |
877 |
6.40 |
|
|
|
| 14 |
A |
636 |
576 |
11.80 |
|
|
|
| 15 |
A |
317 |
287 |
9.60 |
|
|
|
| 16 |
A |
159 |
144 |
0.20 |
|
|
|
| 17 |
A |
122 |
111 |
4.28 |
|
|
|
| 18 |
B |
3307 |
2994 |
58.21 |
|
|
|
| 19 |
B |
3275 |
2966 |
54.67 |
|
|
|
| 20 |
B |
3261 |
2953 |
6.82 |
|
|
|
| 21 |
B |
3190 |
2889 |
83.79 |
|
|
|
| 22 |
B |
1695 |
1535 |
6.79 |
|
|
|
| 23 |
B |
1670 |
1513 |
2.32 |
|
|
|
| 24 |
B |
1635 |
1480 |
4.09 |
|
|
|
| 25 |
B |
1555 |
1408 |
27.33 |
|
|
|
| 26 |
B |
1345 |
1218 |
9.01 |
|
|
|
| 27 |
B |
1270 |
1151 |
13.15 |
|
|
|
| 28 |
B |
1259 |
1140 |
216.00 |
|
|
|
| 29 |
B |
1160 |
1051 |
118.06 |
|
|
|
| 30 |
B |
988 |
895 |
49.73 |
|
|
|
| 31 |
B |
452 |
410 |
10.81 |
|
|
|
| 32 |
B |
218 |
198 |
31.94 |
|
|
|
| 33 |
B |
149 |
135 |
4.39 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 26760.7 cm
-1
Scaled (by 0.9056) Zero Point Vibrational Energy (zpe) 24234.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/3-21G
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.979 |
| H2 |
-0.710 |
0.534 |
1.593 |
| H3 |
0.710 |
-0.534 |
1.593 |
| O4 |
0.772 |
0.887 |
0.192 |
| O5 |
-0.772 |
-0.887 |
0.192 |
| C6 |
0.000 |
1.807 |
-0.607 |
| C7 |
0.000 |
-1.807 |
-0.607 |
| H8 |
0.707 |
2.392 |
-1.173 |
| H9 |
-0.707 |
-2.392 |
-1.173 |
| H10 |
-0.663 |
1.276 |
-1.275 |
| H11 |
-0.590 |
2.463 |
0.025 |
| H12 |
0.663 |
-1.276 |
-1.275 |
| H13 |
0.590 |
-2.463 |
0.025 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
H3 |
O4 |
O5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.0805 | 1.0805 | 1.4154 | 1.4154 | 2.4041 | 2.4041 | 3.2944 | 3.2944 | 2.6731 | 2.7065 | 2.6731 | 2.7065 |
H2 | 1.0805 | | 1.7779 | 2.0704 | 1.9975 | 2.6390 | 3.2904 | 3.6211 | 4.0270 | 2.9624 | 2.4887 | 3.6588 | 3.6243 | H3 | 1.0805 | 1.7779 | | 1.9975 | 2.0704 | 3.2904 | 2.6390 | 4.0270 | 3.6211 | 3.6588 | 3.6243 | 2.9624 | 2.4887 | O4 | 1.4154 | 2.0704 | 1.9975 | | 2.3526 | 1.4420 | 2.9144 | 2.0323 | 3.8479 | 2.0881 | 2.0894 | 2.6155 | 3.3598 | O5 | 1.4154 | 1.9975 | 2.0704 | 2.3526 | | 2.9144 | 1.4420 | 3.8479 | 2.0323 | 2.6155 | 3.3598 | 2.0881 | 2.0894 | C6 | 2.4041 | 2.6390 | 3.2904 | 1.4420 | 2.9144 | | 3.6139 | 1.0786 | 4.2957 | 1.0807 | 1.0854 | 3.2229 | 4.3569 | C7 | 2.4041 | 3.2904 | 2.6390 | 2.9144 | 1.4420 | 3.6139 | | 4.2957 | 1.0786 | 3.2229 | 4.3569 | 1.0807 | 1.0854 | H8 | 3.2944 | 3.6211 | 4.0270 | 2.0323 | 3.8479 | 1.0786 | 4.2957 | | 4.9890 | 1.7706 | 1.7673 | 3.6693 | 5.0024 | H9 | 3.2944 | 4.0270 | 3.6211 | 3.8479 | 2.0323 | 4.2957 | 1.0786 | 4.9890 | | 3.6693 | 5.0024 | 1.7706 | 1.7673 | H10 | 2.6731 | 2.9624 | 3.6588 | 2.0881 | 2.6155 | 1.0807 | 3.2229 | 1.7706 | 3.6693 | | 1.7622 | 2.8751 | 4.1518 | H11 | 2.7065 | 2.4887 | 3.6243 | 2.0894 | 3.3598 | 1.0854 | 4.3569 | 1.7673 | 5.0024 | 1.7622 | | 4.1518 | 5.0658 | H12 | 2.6731 | 3.6588 | 2.9624 | 2.6155 | 2.0881 | 3.2229 | 1.0807 | 3.6693 | 1.7706 | 2.8751 | 4.1518 | | 1.7622 | H13 | 2.7065 | 3.6243 | 2.4887 | 3.3598 | 2.0894 | 4.3569 | 1.0854 | 5.0024 | 1.7673 | 4.1518 | 5.0658 | 1.7622 | |
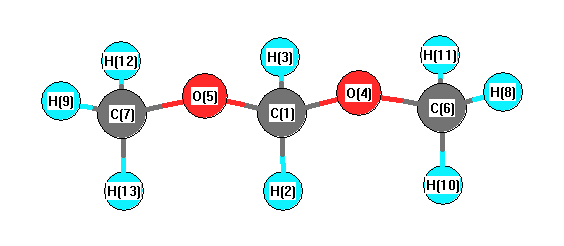 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C6 |
114.566 |
|
C1 |
O5 |
C7 |
114.566 |
| H2 |
C1 |
H3 |
110.710 |
|
H2 |
C1 |
O4 |
111.388 |
| H2 |
C1 |
O5 |
105.527 |
|
H3 |
C1 |
O4 |
105.527 |
| H3 |
C1 |
O5 |
111.388 |
|
O4 |
C1 |
O5 |
112.415 |
| O4 |
C6 |
H8 |
106.586 |
|
O4 |
C6 |
H10 |
110.923 |
| O4 |
C6 |
H11 |
110.742 |
|
O5 |
C7 |
H9 |
106.586 |
| O5 |
C7 |
H12 |
110.923 |
|
O5 |
C7 |
H13 |
110.742 |
| H8 |
C6 |
H10 |
110.165 |
|
H8 |
C6 |
H11 |
109.513 |
| H9 |
C7 |
H12 |
110.165 |
|
H9 |
C7 |
H13 |
109.513 |
| H10 |
C6 |
H11 |
108.889 |
|
H12 |
C7 |
H13 |
108.889 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.267 |
|
|
|
| 2 |
H |
0.202 |
|
|
|
| 3 |
H |
0.202 |
|
|
|
| 4 |
O |
-0.670 |
|
|
|
| 5 |
O |
-0.670 |
|
|
|
| 6 |
C |
-0.272 |
|
|
|
| 7 |
C |
-0.272 |
|
|
|
| 8 |
H |
0.215 |
|
|
|
| 9 |
H |
0.215 |
|
|
|
| 10 |
H |
0.207 |
|
|
|
| 11 |
H |
0.184 |
|
|
|
| 12 |
H |
0.207 |
|
|
|
| 13 |
H |
0.184 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.391 |
0.391 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.561 |
-3.570 |
0.000 |
| y |
-3.570 |
-27.252 |
0.000 |
| z |
0.000 |
0.000 |
-28.419 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.725 |
-3.570 |
0.000 |
| y |
-3.570 |
4.738 |
0.000 |
| z |
0.000 |
0.000 |
2.987 |
|
| Polar |
|---|
| 3z2-r2 | 5.975 |
|---|
| x2-y2 | -8.309 |
|---|
| xy | -3.570 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.421 |
-0.062 |
0.000 |
| y |
-0.062 |
5.872 |
0.000 |
| z |
0.000 |
0.000 |
5.068 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |
Jump to
S1C1
Energy calculated at HF/3-21G
| | hartrees |
|---|
| Energy at 0K | -266.459021 |
| Energy at 298.15K | -266.468539 |
| Nuclear repulsion energy | 190.817818 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3308 |
2996 |
29.19 |
|
|
|
| 2 |
A1 |
3176 |
2876 |
46.52 |
|
|
|
| 3 |
A1 |
3114 |
2820 |
39.91 |
|
|
|
| 4 |
A1 |
1739 |
1575 |
3.51 |
|
|
|
| 5 |
A1 |
1691 |
1531 |
1.75 |
|
|
|
| 6 |
A1 |
1629 |
1475 |
1.49 |
|
|
|
| 7 |
A1 |
1344 |
1217 |
7.42 |
|
|
|
| 8 |
A1 |
1218 |
1103 |
0.18 |
|
|
|
| 9 |
A1 |
1052 |
953 |
61.60 |
|
|
|
| 10 |
A1 |
480 |
435 |
0.37 |
|
|
|
| 11 |
A1 |
216 |
195 |
4.14 |
|
|
|
| 12 |
A2 |
3216 |
2912 |
0.00 |
|
|
|
| 13 |
A2 |
1676 |
1518 |
0.00 |
|
|
|
| 14 |
A2 |
1397 |
1265 |
0.00 |
|
|
|
| 15 |
A2 |
1269 |
1150 |
0.00 |
|
|
|
| 16 |
A2 |
223 |
202 |
0.00 |
|
|
|
| 17 |
A2 |
87 |
78 |
0.00 |
|
|
|
| 18 |
B1 |
3217 |
2913 |
147.51 |
|
|
|
| 19 |
B1 |
3133 |
2837 |
120.58 |
|
|
|
| 20 |
B1 |
1677 |
1518 |
15.98 |
|
|
|
| 21 |
B1 |
1285 |
1163 |
21.32 |
|
|
|
| 22 |
B1 |
1233 |
1116 |
12.84 |
|
|
|
| 23 |
B1 |
246 |
223 |
11.76 |
|
|
|
| 24 |
B1 |
78 |
70 |
1.70 |
|
|
|
| 25 |
B2 |
3308 |
2995 |
24.11 |
|
|
|
| 26 |
B2 |
3174 |
2875 |
42.23 |
|
|
|
| 27 |
B2 |
1696 |
1536 |
2.17 |
|
|
|
| 28 |
B2 |
1660 |
1503 |
36.67 |
|
|
|
| 29 |
B2 |
1590 |
1440 |
107.65 |
|
|
|
| 30 |
B2 |
1310 |
1186 |
203.44 |
|
|
|
| 31 |
B2 |
1235 |
1118 |
231.29 |
|
|
|
| 32 |
B2 |
1034 |
936 |
8.45 |
|
|
|
| 33 |
B2 |
422 |
382 |
4.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 26564.8 cm
-1
Scaled (by 0.9056) Zero Point Vibrational Energy (zpe) 24057.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/3-21G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.334 |
| H2 |
-0.881 |
0.000 |
0.975 |
| H3 |
0.881 |
0.000 |
0.975 |
| O4 |
0.000 |
1.129 |
-0.511 |
| O5 |
0.000 |
-1.129 |
-0.511 |
| C6 |
0.000 |
2.387 |
0.181 |
| C7 |
0.000 |
-2.387 |
0.181 |
| H8 |
0.000 |
3.152 |
-0.579 |
| H9 |
0.000 |
-3.152 |
-0.579 |
| H10 |
-0.882 |
2.500 |
0.803 |
| H11 |
0.882 |
2.500 |
0.803 |
| H12 |
0.882 |
-2.500 |
0.803 |
| H13 |
-0.882 |
-2.500 |
0.803 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
H3 |
O4 |
O5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.0899 | 1.0899 | 1.4101 | 1.4101 | 2.3921 | 2.3921 | 3.2814 | 3.2814 | 2.6919 | 2.6919 | 2.6919 | 2.6919 |
H2 | 1.0899 | | 1.7623 | 2.0639 | 2.0639 | 2.6658 | 2.6658 | 3.6230 | 3.6230 | 2.5054 | 3.0639 | 3.0639 | 2.5054 | H3 | 1.0899 | 1.7623 | | 2.0639 | 2.0639 | 2.6658 | 2.6658 | 3.6230 | 3.6230 | 3.0639 | 2.5054 | 2.5054 | 3.0639 | O4 | 1.4101 | 2.0639 | 2.0639 | | 2.2584 | 1.4355 | 3.5837 | 2.0238 | 4.2816 | 2.0935 | 2.0935 | 3.9588 | 3.9588 | O5 | 1.4101 | 2.0639 | 2.0639 | 2.2584 | | 3.5837 | 1.4355 | 4.2816 | 2.0238 | 3.9588 | 3.9588 | 2.0935 | 2.0935 | C6 | 2.3921 | 2.6658 | 2.6658 | 1.4355 | 3.5837 | | 4.7744 | 1.0779 | 5.5909 | 1.0857 | 1.0857 | 5.0046 | 5.0046 | C7 | 2.3921 | 2.6658 | 2.6658 | 3.5837 | 1.4355 | 4.7744 | | 5.5909 | 1.0779 | 5.0046 | 5.0046 | 1.0857 | 1.0857 | H8 | 3.2814 | 3.6230 | 3.6230 | 2.0238 | 4.2816 | 1.0779 | 5.5909 | | 6.3037 | 1.7648 | 1.7648 | 5.8844 | 5.8844 | H9 | 3.2814 | 3.6230 | 3.6230 | 4.2816 | 2.0238 | 5.5909 | 1.0779 | 6.3037 | | 5.8844 | 5.8844 | 1.7648 | 1.7648 | H10 | 2.6919 | 2.5054 | 3.0639 | 2.0935 | 3.9588 | 1.0857 | 5.0046 | 1.7648 | 5.8844 | | 1.7650 | 5.3014 | 4.9990 | H11 | 2.6919 | 3.0639 | 2.5054 | 2.0935 | 3.9588 | 1.0857 | 5.0046 | 1.7648 | 5.8844 | 1.7650 | | 4.9990 | 5.3014 | H12 | 2.6919 | 3.0639 | 2.5054 | 3.9588 | 2.0935 | 5.0046 | 1.0857 | 5.8844 | 1.7648 | 5.3014 | 4.9990 | | 1.7650 | H13 | 2.6919 | 2.5054 | 3.0639 | 3.9588 | 2.0935 | 5.0046 | 1.0857 | 5.8844 | 1.7648 | 4.9990 | 5.3014 | 1.7650 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C6 |
114.409 |
|
C1 |
O5 |
C7 |
114.409 |
| H2 |
C1 |
H3 |
107.894 |
|
H2 |
C1 |
O4 |
110.642 |
| H2 |
C1 |
O5 |
110.642 |
|
H3 |
C1 |
O4 |
110.642 |
| H3 |
C1 |
O5 |
110.642 |
|
O4 |
C1 |
O5 |
106.406 |
| O4 |
C6 |
H8 |
106.396 |
|
O4 |
C6 |
H10 |
111.518 |
| O4 |
C6 |
H11 |
111.518 |
|
O5 |
C7 |
H9 |
106.396 |
| O5 |
C7 |
H12 |
111.518 |
|
O5 |
C7 |
H13 |
111.518 |
| H8 |
C6 |
H10 |
109.306 |
|
H8 |
C6 |
H11 |
109.306 |
| H9 |
C7 |
H12 |
109.306 |
|
H9 |
C7 |
H13 |
109.306 |
| H10 |
C6 |
H11 |
108.742 |
|
H12 |
C7 |
H13 |
108.742 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.337 |
|
|
|
| 2 |
H |
0.158 |
|
|
|
| 3 |
H |
0.158 |
|
|
|
| 4 |
O |
-0.651 |
|
|
|
| 5 |
O |
-0.651 |
|
|
|
| 6 |
C |
-0.270 |
|
|
|
| 7 |
C |
-0.270 |
|
|
|
| 8 |
H |
0.230 |
|
|
|
| 9 |
H |
0.230 |
|
|
|
| 10 |
H |
0.183 |
|
|
|
| 11 |
H |
0.183 |
|
|
|
| 12 |
H |
0.183 |
|
|
|
| 13 |
H |
0.183 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.509 |
3.509 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.005 |
0.000 |
0.000 |
| y |
0.000 |
-22.545 |
0.000 |
| z |
0.000 |
0.000 |
-33.701 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.882 |
0.000 |
0.000 |
| y |
0.000 |
10.308 |
0.000 |
| z |
0.000 |
0.000 |
-6.426 |
|
| Polar |
|---|
| 3z2-r2 | -12.852 |
|---|
| x2-y2 | -9.460 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.595 |
0.000 |
0.000 |
| y |
0.000 |
6.881 |
0.000 |
| z |
0.000 |
0.000 |
4.512 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |