Jump to
S1C2
Energy calculated at HF/3-21G
| | hartrees |
|---|
| Energy at 0K | -308.501521 |
| Energy at 298.15K | -308.517428 |
| HF Energy | -308.501521 |
| Nuclear repulsion energy | 313.428784 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3259 |
2952 |
63.11 |
|
|
|
| 2 |
A1 |
3229 |
2924 |
22.66 |
|
|
|
| 3 |
A1 |
3193 |
2892 |
26.38 |
|
|
|
| 4 |
A1 |
3168 |
2869 |
60.78 |
|
|
|
| 5 |
A1 |
1711 |
1550 |
0.18 |
|
|
|
| 6 |
A1 |
1676 |
1517 |
9.22 |
|
|
|
| 7 |
A1 |
1663 |
1506 |
3.54 |
|
|
|
| 8 |
A1 |
1594 |
1443 |
0.07 |
|
|
|
| 9 |
A1 |
1572 |
1424 |
5.11 |
|
|
|
| 10 |
A1 |
1479 |
1339 |
0.39 |
|
|
|
| 11 |
A1 |
1260 |
1141 |
0.15 |
|
|
|
| 12 |
A1 |
1094 |
991 |
4.08 |
|
|
|
| 13 |
A1 |
1029 |
932 |
21.50 |
|
|
|
| 14 |
A1 |
981 |
889 |
4.05 |
|
|
|
| 15 |
A1 |
453 |
411 |
1.39 |
|
|
|
| 16 |
A1 |
332 |
301 |
0.11 |
|
|
|
| 17 |
A1 |
106 |
96 |
0.31 |
|
|
|
| 18 |
A2 |
3277 |
2968 |
0.00 |
|
|
|
| 19 |
A2 |
3238 |
2933 |
0.00 |
|
|
|
| 20 |
A2 |
3187 |
2886 |
0.00 |
|
|
|
| 21 |
A2 |
1675 |
1517 |
0.00 |
|
|
|
| 22 |
A2 |
1457 |
1319 |
0.00 |
|
|
|
| 23 |
A2 |
1394 |
1262 |
0.00 |
|
|
|
| 24 |
A2 |
1285 |
1164 |
0.00 |
|
|
|
| 25 |
A2 |
998 |
904 |
0.00 |
|
|
|
| 26 |
A2 |
844 |
764 |
0.00 |
|
|
|
| 27 |
A2 |
251 |
227 |
0.00 |
|
|
|
| 28 |
A2 |
141 |
128 |
0.00 |
|
|
|
| 29 |
A2 |
80 |
73 |
0.00 |
|
|
|
| 30 |
B1 |
3278 |
2968 |
119.19 |
|
|
|
| 31 |
B1 |
3238 |
2933 |
20.10 |
|
|
|
| 32 |
B1 |
3192 |
2891 |
92.72 |
|
|
|
| 33 |
B1 |
1675 |
1517 |
17.52 |
|
|
|
| 34 |
B1 |
1459 |
1321 |
0.00 |
|
|
|
| 35 |
B1 |
1398 |
1266 |
2.91 |
|
|
|
| 36 |
B1 |
1307 |
1184 |
5.31 |
|
|
|
| 37 |
B1 |
1012 |
917 |
4.75 |
|
|
|
| 38 |
B1 |
846 |
766 |
4.94 |
|
|
|
| 39 |
B1 |
252 |
228 |
0.24 |
|
|
|
| 40 |
B1 |
170 |
154 |
10.06 |
|
|
|
| 41 |
B1 |
52 |
47 |
0.75 |
|
|
|
| 42 |
B2 |
3259 |
2951 |
19.11 |
|
|
|
| 43 |
B2 |
3229 |
2924 |
6.01 |
|
|
|
| 44 |
B2 |
3193 |
2891 |
41.04 |
|
|
|
| 45 |
B2 |
3159 |
2860 |
4.51 |
|
|
|
| 46 |
B2 |
1696 |
1536 |
10.27 |
|
|
|
| 47 |
B2 |
1675 |
1517 |
2.05 |
|
|
|
| 48 |
B2 |
1661 |
1505 |
1.26 |
|
|
|
| 49 |
B2 |
1577 |
1428 |
16.73 |
|
|
|
| 50 |
B2 |
1526 |
1382 |
51.32 |
|
|
|
| 51 |
B2 |
1465 |
1327 |
13.75 |
|
|
|
| 52 |
B2 |
1236 |
1119 |
237.63 |
|
|
|
| 53 |
B2 |
1223 |
1108 |
0.01 |
|
|
|
| 54 |
B2 |
1071 |
970 |
0.15 |
|
|
|
| 55 |
B2 |
949 |
859 |
4.50 |
|
|
|
| 56 |
B2 |
533 |
483 |
7.38 |
|
|
|
| 57 |
B2 |
267 |
241 |
2.16 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 45611.6 cm
-1
Scaled (by 0.9056) Zero Point Vibrational Energy (zpe) 41305.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/3-21G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.318 |
| C2 |
0.000 |
1.215 |
-0.447 |
| C3 |
0.000 |
-1.215 |
-0.447 |
| C4 |
0.000 |
2.373 |
0.547 |
| C5 |
0.000 |
-2.373 |
0.547 |
| C6 |
0.000 |
3.735 |
-0.169 |
| C7 |
0.000 |
-3.735 |
-0.169 |
| H8 |
0.879 |
1.262 |
-1.084 |
| H9 |
-0.879 |
1.262 |
-1.084 |
| H10 |
-0.879 |
-1.262 |
-1.084 |
| H11 |
0.879 |
-1.262 |
-1.084 |
| H12 |
0.874 |
2.276 |
1.179 |
| H13 |
-0.874 |
2.276 |
1.179 |
| H14 |
-0.874 |
-2.276 |
1.179 |
| H15 |
0.874 |
-2.276 |
1.179 |
| H16 |
0.000 |
4.548 |
0.548 |
| H17 |
-0.879 |
3.842 |
-0.797 |
| H18 |
0.879 |
3.842 |
-0.797 |
| H19 |
0.000 |
-4.548 |
0.548 |
| H20 |
0.879 |
-3.842 |
-0.797 |
| H21 |
-0.879 |
-3.842 |
-0.797 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4351 | 1.4351 | 2.3838 | 2.3838 | 3.7667 | 3.7667 | 2.0813 | 2.0813 | 2.0813 | 2.0813 | 2.5855 | 2.5855 | 2.5855 | 2.5855 | 4.5535 | 4.0954 | 4.0954 | 4.5535 | 4.0954 | 4.0954 |
C2 | 1.4351 | | 2.4291 | 1.5262 | 3.7224 | 2.5359 | 4.9575 | 1.0871 | 1.0871 | 2.7045 | 2.7045 | 2.1294 | 2.1294 | 3.9484 | 3.9484 | 3.4784 | 2.7923 | 2.7923 | 5.8474 | 5.1440 | 5.1440 | C3 | 1.4351 | 2.4291 | | 3.7224 | 1.5262 | 4.9575 | 2.5359 | 2.7045 | 2.7045 | 1.0871 | 1.0871 | 3.9484 | 3.9484 | 2.1294 | 2.1294 | 5.8474 | 5.1440 | 5.1440 | 3.4784 | 2.7923 | 2.7923 | C4 | 2.3838 | 1.5262 | 3.7224 | | 4.7455 | 1.5392 | 6.1498 | 2.1604 | 2.1604 | 4.0802 | 4.0802 | 1.0829 | 1.0829 | 4.7720 | 4.7720 | 2.1749 | 2.1761 | 2.1761 | 6.9204 | 6.4185 | 6.4185 | C5 | 2.3838 | 3.7224 | 1.5262 | 4.7455 | | 6.1498 | 1.5392 | 4.0802 | 4.0802 | 2.1604 | 2.1604 | 4.7720 | 4.7720 | 1.0829 | 1.0829 | 6.9204 | 6.4185 | 6.4185 | 2.1749 | 2.1761 | 2.1761 | C6 | 3.7667 | 2.5359 | 4.9575 | 1.5392 | 6.1498 | | 7.4703 | 2.7794 | 2.7794 | 5.1562 | 5.1562 | 2.1707 | 2.1707 | 6.2220 | 6.2220 | 1.0837 | 1.0852 | 1.0852 | 8.3138 | 7.6534 | 7.6534 | C7 | 3.7667 | 4.9575 | 2.5359 | 6.1498 | 1.5392 | 7.4703 | | 5.1562 | 5.1562 | 2.7794 | 2.7794 | 6.2220 | 6.2220 | 2.1707 | 2.1707 | 8.3138 | 7.6534 | 7.6534 | 1.0837 | 1.0852 | 1.0852 | H8 | 2.0813 | 1.0871 | 2.7045 | 2.1604 | 4.0802 | 2.7794 | 5.1562 | | 1.7587 | 3.0769 | 2.5248 | 2.4800 | 3.0371 | 4.5514 | 4.2002 | 3.7723 | 3.1349 | 2.5953 | 6.0987 | 5.1122 | 5.4062 | H9 | 2.0813 | 1.0871 | 2.7045 | 2.1604 | 4.0802 | 2.7794 | 5.1562 | 1.7587 | | 2.5248 | 3.0769 | 3.0371 | 2.4800 | 4.2002 | 4.5514 | 3.7723 | 2.5953 | 3.1349 | 6.0987 | 5.4062 | 5.1122 | H10 | 2.0813 | 2.7045 | 1.0871 | 4.0802 | 2.1604 | 5.1562 | 2.7794 | 3.0769 | 2.5248 | | 1.7587 | 4.5514 | 4.2002 | 2.4800 | 3.0371 | 6.0987 | 5.1122 | 5.4062 | 3.7723 | 3.1349 | 2.5953 | H11 | 2.0813 | 2.7045 | 1.0871 | 4.0802 | 2.1604 | 5.1562 | 2.7794 | 2.5248 | 3.0769 | 1.7587 | | 4.2002 | 4.5514 | 3.0371 | 2.4800 | 6.0987 | 5.4062 | 5.1122 | 3.7723 | 2.5953 | 3.1349 | H12 | 2.5855 | 2.1294 | 3.9484 | 1.0829 | 4.7720 | 2.1707 | 6.2220 | 2.4800 | 3.0371 | 4.5514 | 4.2002 | | 1.7476 | 4.8754 | 4.5515 | 2.5147 | 3.0706 | 2.5211 | 6.9080 | 6.4286 | 6.6633 | H13 | 2.5855 | 2.1294 | 3.9484 | 1.0829 | 4.7720 | 2.1707 | 6.2220 | 3.0371 | 2.4800 | 4.2002 | 4.5514 | 1.7476 | | 4.5515 | 4.8754 | 2.5147 | 2.5211 | 3.0706 | 6.9080 | 6.6633 | 6.4286 | H14 | 2.5855 | 3.9484 | 2.1294 | 4.7720 | 1.0829 | 6.2220 | 2.1707 | 4.5514 | 4.2002 | 2.4800 | 3.0371 | 4.8754 | 4.5515 | | 1.7476 | 6.9080 | 6.4286 | 6.6633 | 2.5147 | 3.0706 | 2.5211 | H15 | 2.5855 | 3.9484 | 2.1294 | 4.7720 | 1.0829 | 6.2220 | 2.1707 | 4.2002 | 4.5514 | 3.0371 | 2.4800 | 4.5515 | 4.8754 | 1.7476 | | 6.9080 | 6.6633 | 6.4286 | 2.5147 | 2.5211 | 3.0706 | H16 | 4.5535 | 3.4784 | 5.8474 | 2.1749 | 6.9204 | 1.0837 | 8.3138 | 3.7723 | 3.7723 | 6.0987 | 6.0987 | 2.5147 | 2.5147 | 6.9080 | 6.9080 | | 1.7546 | 1.7546 | 9.0953 | 8.5417 | 8.5417 | H17 | 4.0954 | 2.7923 | 5.1440 | 2.1761 | 6.4185 | 1.0852 | 7.6534 | 3.1349 | 2.5953 | 5.1122 | 5.4062 | 3.0706 | 2.5211 | 6.4286 | 6.6633 | 1.7546 | | 1.7581 | 8.5417 | 7.8820 | 7.6834 | H18 | 4.0954 | 2.7923 | 5.1440 | 2.1761 | 6.4185 | 1.0852 | 7.6534 | 2.5953 | 3.1349 | 5.4062 | 5.1122 | 2.5211 | 3.0706 | 6.6633 | 6.4286 | 1.7546 | 1.7581 | | 8.5417 | 7.6834 | 7.8820 | H19 | 4.5535 | 5.8474 | 3.4784 | 6.9204 | 2.1749 | 8.3138 | 1.0837 | 6.0987 | 6.0987 | 3.7723 | 3.7723 | 6.9080 | 6.9080 | 2.5147 | 2.5147 | 9.0953 | 8.5417 | 8.5417 | | 1.7546 | 1.7546 | H20 | 4.0954 | 5.1440 | 2.7923 | 6.4185 | 2.1761 | 7.6534 | 1.0852 | 5.1122 | 5.4062 | 3.1349 | 2.5953 | 6.4286 | 6.6633 | 3.0706 | 2.5211 | 8.5417 | 7.8820 | 7.6834 | 1.7546 | | 1.7581 | H21 | 4.0954 | 5.1440 | 2.7923 | 6.4185 | 2.1761 | 7.6534 | 1.0852 | 5.4062 | 5.1122 | 2.5953 | 3.1349 | 6.6633 | 6.4286 | 2.5211 | 3.0706 | 8.5417 | 7.6834 | 7.8820 | 1.7546 | 1.7581 | |
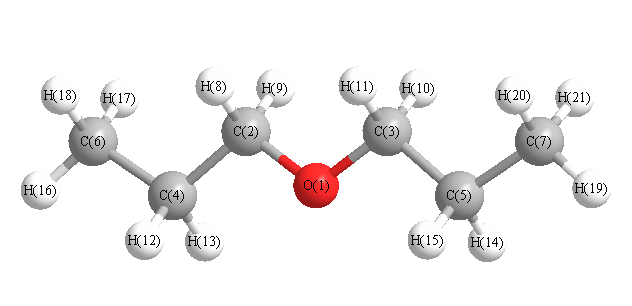 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
107.178 |
|
O1 |
C2 |
H8 |
110.459 |
| O1 |
C2 |
H9 |
110.459 |
|
O1 |
C3 |
C5 |
107.178 |
| O1 |
C3 |
H10 |
110.459 |
|
O1 |
C3 |
H11 |
110.459 |
| C2 |
O1 |
C3 |
115.626 |
|
C2 |
C4 |
C6 |
111.635 |
| C2 |
C4 |
H12 |
108.194 |
|
C2 |
C4 |
H13 |
108.194 |
| C3 |
C5 |
C7 |
111.635 |
|
C3 |
C5 |
H14 |
108.194 |
| C3 |
C5 |
H15 |
108.194 |
|
C4 |
C2 |
H8 |
110.391 |
| C4 |
C2 |
H9 |
110.391 |
|
C4 |
C6 |
H16 |
110.835 |
| C4 |
C6 |
H17 |
110.848 |
|
C4 |
C6 |
H18 |
110.848 |
| C5 |
C3 |
H10 |
110.391 |
|
C5 |
C3 |
H11 |
110.391 |
| C5 |
C7 |
H19 |
110.835 |
|
C5 |
C7 |
H20 |
110.848 |
| C5 |
C7 |
H21 |
110.848 |
|
C6 |
C4 |
H12 |
110.548 |
| C6 |
C4 |
H13 |
110.548 |
|
C7 |
C5 |
H14 |
110.548 |
| C7 |
C5 |
H15 |
110.548 |
|
H8 |
C2 |
H9 |
107.979 |
| H10 |
C3 |
H11 |
107.979 |
|
H12 |
C4 |
H13 |
107.586 |
| H14 |
C5 |
H15 |
107.586 |
|
H16 |
C6 |
H17 |
107.994 |
| H16 |
C6 |
H18 |
107.994 |
|
H17 |
C6 |
H18 |
108.207 |
| H19 |
C7 |
H20 |
107.994 |
|
H19 |
C7 |
H21 |
107.994 |
| H20 |
C7 |
H21 |
108.207 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.661 |
|
|
|
| 2 |
C |
-0.030 |
|
|
|
| 3 |
C |
-0.030 |
|
|
|
| 4 |
C |
-0.455 |
|
|
|
| 5 |
C |
-0.455 |
|
|
|
| 6 |
C |
-0.601 |
|
|
|
| 7 |
C |
-0.601 |
|
|
|
| 8 |
H |
0.180 |
|
|
|
| 9 |
H |
0.180 |
|
|
|
| 10 |
H |
0.180 |
|
|
|
| 11 |
H |
0.180 |
|
|
|
| 12 |
H |
0.225 |
|
|
|
| 13 |
H |
0.225 |
|
|
|
| 14 |
H |
0.225 |
|
|
|
| 15 |
H |
0.225 |
|
|
|
| 16 |
H |
0.209 |
|
|
|
| 17 |
H |
0.198 |
|
|
|
| 18 |
H |
0.198 |
|
|
|
| 19 |
H |
0.209 |
|
|
|
| 20 |
H |
0.198 |
|
|
|
| 21 |
H |
0.198 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-1.368 |
1.368 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.549 |
0.000 |
0.000 |
| y |
0.000 |
-43.984 |
0.000 |
| z |
0.000 |
0.000 |
-47.275 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.920 |
0.000 |
0.000 |
| y |
0.000 |
2.929 |
0.000 |
| z |
0.000 |
0.000 |
-2.008 |
|
| Polar |
|---|
| 3z2-r2 | -4.017 |
|---|
| x2-y2 | -2.566 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.319 |
0.000 |
0.000 |
| y |
0.000 |
11.544 |
0.000 |
| z |
0.000 |
0.000 |
8.427 |
<r2> (average value of r
2) Å
2
| <r2> |
438.635 |
| (<r2>)1/2 |
20.944 |
Jump to
S1C1
Energy calculated at HF/3-21G
| | hartrees |
|---|
| Energy at 0K | -308.503662 |
| Energy at 298.15K | -308.519874 |
| HF Energy | -308.503662 |
| Nuclear repulsion energy | 326.208398 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3300 |
2988 |
7.29 |
|
|
|
| 2 |
A |
3262 |
2954 |
58.13 |
|
|
|
| 3 |
A |
3243 |
2936 |
10.75 |
|
|
|
| 4 |
A |
3216 |
2912 |
13.40 |
|
|
|
| 5 |
A |
3197 |
2896 |
6.15 |
|
|
|
| 6 |
A |
3193 |
2892 |
0.03 |
|
|
|
| 7 |
A |
3172 |
2873 |
76.61 |
|
|
|
| 8 |
A |
1709 |
1548 |
0.88 |
|
|
|
| 9 |
A |
1684 |
1525 |
6.35 |
|
|
|
| 10 |
A |
1668 |
1511 |
7.99 |
|
|
|
| 11 |
A |
1647 |
1492 |
0.43 |
|
|
|
| 12 |
A |
1592 |
1442 |
3.83 |
|
|
|
| 13 |
A |
1563 |
1415 |
3.19 |
|
|
|
| 14 |
A |
1518 |
1375 |
0.00 |
|
|
|
| 15 |
A |
1447 |
1311 |
1.24 |
|
|
|
| 16 |
A |
1391 |
1260 |
0.05 |
|
|
|
| 17 |
A |
1285 |
1164 |
2.47 |
|
|
|
| 18 |
A |
1242 |
1125 |
9.89 |
|
|
|
| 19 |
A |
1118 |
1012 |
3.18 |
|
|
|
| 20 |
A |
1037 |
939 |
0.00 |
|
|
|
| 21 |
A |
1007 |
912 |
16.09 |
|
|
|
| 22 |
A |
930 |
842 |
0.64 |
|
|
|
| 23 |
A |
822 |
744 |
1.22 |
|
|
|
| 24 |
A |
509 |
461 |
0.08 |
|
|
|
| 25 |
A |
364 |
329 |
1.09 |
|
|
|
| 26 |
A |
287 |
260 |
0.87 |
|
|
|
| 27 |
A |
174 |
158 |
0.12 |
|
|
|
| 28 |
A |
125 |
113 |
0.02 |
|
|
|
| 29 |
A |
51 |
47 |
0.00 |
|
|
|
| 30 |
B |
3300 |
2988 |
39.87 |
|
|
|
| 31 |
B |
3262 |
2954 |
51.94 |
|
|
|
| 32 |
B |
3242 |
2936 |
31.47 |
|
|
|
| 33 |
B |
3216 |
2912 |
77.75 |
|
|
|
| 34 |
B |
3199 |
2897 |
133.53 |
|
|
|
| 35 |
B |
3196 |
2894 |
0.52 |
|
|
|
| 36 |
B |
3163 |
2864 |
2.04 |
|
|
|
| 37 |
B |
1692 |
1533 |
8.33 |
|
|
|
| 38 |
B |
1683 |
1524 |
6.79 |
|
|
|
| 39 |
B |
1668 |
1510 |
5.32 |
|
|
|
| 40 |
B |
1647 |
1492 |
5.87 |
|
|
|
| 41 |
B |
1567 |
1419 |
17.38 |
|
|
|
| 42 |
B |
1522 |
1378 |
46.85 |
|
|
|
| 43 |
B |
1516 |
1373 |
17.59 |
|
|
|
| 44 |
B |
1437 |
1301 |
0.19 |
|
|
|
| 45 |
B |
1396 |
1264 |
6.18 |
|
|
|
| 46 |
B |
1289 |
1167 |
4.79 |
|
|
|
| 47 |
B |
1248 |
1130 |
98.88 |
|
|
|
| 48 |
B |
1198 |
1085 |
118.15 |
|
|
|
| 49 |
B |
1088 |
985 |
17.74 |
|
|
|
| 50 |
B |
1031 |
934 |
0.86 |
|
|
|
| 51 |
B |
910 |
824 |
0.58 |
|
|
|
| 52 |
B |
867 |
785 |
5.77 |
|
|
|
| 53 |
B |
531 |
481 |
1.68 |
|
|
|
| 54 |
B |
342 |
310 |
1.54 |
|
|
|
| 55 |
B |
254 |
230 |
5.90 |
|
|
|
| 56 |
B |
189 |
171 |
8.12 |
|
|
|
| 57 |
B |
73 |
67 |
2.64 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 45738.1 cm
-1
Scaled (by 0.9056) Zero Point Vibrational Energy (zpe) 41420.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/3-21G
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.175 |
| C2 |
0.028 |
1.216 |
0.938 |
| C3 |
-0.028 |
-1.216 |
0.938 |
| C4 |
0.000 |
2.371 |
-0.061 |
| C5 |
0.000 |
-2.371 |
-0.061 |
| C6 |
-1.276 |
2.328 |
-0.919 |
| C7 |
1.276 |
-2.328 |
-0.919 |
| H8 |
-0.835 |
1.267 |
1.597 |
| H9 |
0.925 |
1.254 |
1.550 |
| H10 |
0.835 |
-1.267 |
1.597 |
| H11 |
-0.925 |
-1.254 |
1.550 |
| H12 |
0.873 |
2.287 |
-0.697 |
| H13 |
0.064 |
3.310 |
0.479 |
| H14 |
-0.873 |
-2.287 |
-0.697 |
| H15 |
-0.064 |
-3.310 |
0.479 |
| H16 |
-1.267 |
3.108 |
-1.672 |
| H17 |
-1.343 |
1.366 |
-1.407 |
| H18 |
-2.157 |
2.463 |
-0.300 |
| H19 |
1.267 |
-3.108 |
-1.672 |
| H20 |
1.343 |
-1.366 |
-1.407 |
| H21 |
2.157 |
-2.463 |
-0.300 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4363 | 1.4363 | 2.3829 | 2.3829 | 2.8708 | 2.8708 | 2.0798 | 2.0785 | 2.0798 | 2.0785 | 2.5987 | 3.3248 | 2.5987 | 3.3248 | 3.8305 | 2.4837 | 3.3082 | 3.8305 | 2.4837 | 3.3082 |
C2 | 1.4363 | | 2.4331 | 1.5272 | 3.7241 | 2.5267 | 4.1912 | 1.0869 | 1.0864 | 2.6927 | 2.7176 | 2.1294 | 2.1439 | 3.9698 | 4.5506 | 3.4738 | 2.7203 | 2.8037 | 5.2005 | 3.7275 | 4.4277 | C3 | 1.4363 | 2.4331 | | 3.7241 | 1.5272 | 4.1912 | 2.5267 | 2.6927 | 2.7176 | 1.0869 | 1.0864 | 3.9698 | 4.5506 | 2.1294 | 2.1439 | 5.2005 | 3.7275 | 4.4277 | 3.4738 | 2.7203 | 2.8037 | C4 | 2.3829 | 1.5272 | 3.7241 | | 4.7426 | 1.5383 | 4.9441 | 2.1601 | 2.1672 | 4.0841 | 4.0736 | 1.0833 | 1.0850 | 4.7822 | 5.7074 | 2.1781 | 2.1510 | 2.1719 | 5.8499 | 4.1928 | 5.2992 | C5 | 2.3829 | 3.7241 | 1.5272 | 4.7426 | | 4.9441 | 1.5383 | 4.0841 | 4.0736 | 2.1601 | 2.1672 | 4.7822 | 5.7074 | 1.0833 | 1.0850 | 5.8499 | 4.1928 | 5.2992 | 2.1781 | 2.1510 | 2.1719 | C6 | 2.8708 | 2.5267 | 4.1912 | 1.5383 | 4.9441 | | 5.3083 | 2.7665 | 3.4772 | 4.8689 | 4.3647 | 2.1602 | 2.1719 | 4.6378 | 5.9337 | 1.0841 | 1.0806 | 1.0855 | 6.0475 | 4.5532 | 5.9258 | C7 | 2.8708 | 4.1912 | 2.5267 | 4.9441 | 1.5383 | 5.3083 | | 4.8689 | 4.3647 | 2.7665 | 3.4772 | 4.6378 | 5.9337 | 2.1602 | 2.1719 | 6.0475 | 4.5532 | 5.9258 | 1.0841 | 1.0806 | 1.0855 | H8 | 2.0798 | 1.0869 | 2.6927 | 2.1601 | 4.0841 | 2.7665 | 4.8689 | | 1.7602 | 3.0341 | 2.5230 | 3.0366 | 2.4969 | 4.2303 | 4.7740 | 3.7769 | 3.0485 | 2.6037 | 5.8516 | 4.5493 | 5.1441 | H9 | 2.0785 | 1.0864 | 2.7176 | 2.1672 | 4.0736 | 3.4772 | 4.3647 | 1.7602 | | 2.5230 | 3.1165 | 2.4735 | 2.4725 | 4.5631 | 4.7915 | 4.3151 | 3.7279 | 3.7921 | 5.4338 | 3.9727 | 4.3314 | H10 | 2.0798 | 2.6927 | 1.0869 | 4.0841 | 2.1601 | 4.8689 | 2.7665 | 3.0341 | 2.5230 | | 1.7602 | 4.2303 | 4.7740 | 3.0366 | 2.4969 | 5.8516 | 4.5493 | 5.1441 | 3.7769 | 3.0485 | 2.6037 | H11 | 2.0785 | 2.7176 | 1.0864 | 4.0736 | 2.1672 | 4.3647 | 3.4772 | 2.5230 | 3.1165 | 1.7602 | | 4.5631 | 4.7915 | 2.4735 | 2.4725 | 5.4338 | 3.9727 | 4.3314 | 4.3151 | 3.7279 | 3.7921 | H12 | 2.5987 | 2.1294 | 3.9698 | 1.0833 | 4.7822 | 2.1602 | 4.6378 | 3.0366 | 2.4735 | 4.2303 | 4.5631 | | 1.7560 | 4.8964 | 5.7960 | 2.4903 | 2.5024 | 3.0604 | 5.4966 | 3.7508 | 4.9369 | H13 | 3.3248 | 2.1439 | 4.5506 | 1.0850 | 5.7074 | 2.1719 | 5.9337 | 2.4969 | 2.4725 | 4.7740 | 4.7915 | 1.7560 | | 5.7960 | 6.6216 | 2.5380 | 3.0528 | 2.5015 | 6.8749 | 5.2015 | 6.1902 | H14 | 2.5987 | 3.9698 | 2.1294 | 4.7822 | 1.0833 | 4.6378 | 2.1602 | 4.2303 | 4.5631 | 3.0366 | 2.4735 | 4.8964 | 5.7960 | | 1.7560 | 5.4966 | 3.7508 | 4.9369 | 2.4903 | 2.5024 | 3.0604 | H15 | 3.3248 | 4.5506 | 2.1439 | 5.7074 | 1.0850 | 5.9337 | 2.1719 | 4.7740 | 4.7915 | 2.4969 | 2.4725 | 5.7960 | 6.6216 | 1.7560 | | 6.8749 | 5.2015 | 6.1902 | 2.5380 | 3.0528 | 2.5015 | H16 | 3.8305 | 3.4738 | 5.2005 | 2.1781 | 5.8499 | 1.0841 | 6.0475 | 3.7769 | 4.3151 | 5.8516 | 5.4338 | 2.4903 | 2.5380 | 5.4966 | 6.8749 | | 1.7639 | 1.7578 | 6.7119 | 5.1854 | 6.6811 | H17 | 2.4837 | 2.7203 | 3.7275 | 2.1510 | 4.1928 | 1.0806 | 4.5532 | 3.0485 | 3.7279 | 4.5493 | 3.9727 | 2.5024 | 3.0528 | 3.7508 | 5.2015 | 1.7639 | | 1.7586 | 5.1854 | 3.8300 | 5.3037 | H18 | 3.3082 | 2.8037 | 4.4277 | 2.1719 | 5.2992 | 1.0855 | 5.9258 | 2.6037 | 3.7921 | 5.1441 | 4.3314 | 3.0604 | 2.5015 | 4.9369 | 6.1902 | 1.7578 | 1.7586 | | 6.6811 | 5.3037 | 6.5479 | H19 | 3.8305 | 5.2005 | 3.4738 | 5.8499 | 2.1781 | 6.0475 | 1.0841 | 5.8516 | 5.4338 | 3.7769 | 4.3151 | 5.4966 | 6.8749 | 2.4903 | 2.5380 | 6.7119 | 5.1854 | 6.6811 | | 1.7639 | 1.7578 | H20 | 2.4837 | 3.7275 | 2.7203 | 4.1928 | 2.1510 | 4.5532 | 1.0806 | 4.5493 | 3.9727 | 3.0485 | 3.7279 | 3.7508 | 5.2015 | 2.5024 | 3.0528 | 5.1854 | 3.8300 | 5.3037 | 1.7639 | | 1.7586 | H21 | 3.3082 | 4.4277 | 2.8037 | 5.2992 | 2.1719 | 5.9258 | 1.0855 | 5.1441 | 4.3314 | 2.6037 | 3.7921 | 4.9369 | 6.1902 | 3.0604 | 2.5015 | 6.6811 | 5.3037 | 6.5479 | 1.7578 | 1.7586 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
107.002 |
|
O1 |
C2 |
H8 |
110.261 |
| O1 |
C2 |
H9 |
110.186 |
|
O1 |
C3 |
C5 |
107.002 |
| O1 |
C3 |
H10 |
110.261 |
|
O1 |
C3 |
H11 |
110.186 |
| C2 |
O1 |
C3 |
115.771 |
|
C2 |
C4 |
C6 |
111.021 |
| C2 |
C4 |
H12 |
108.107 |
|
C2 |
C4 |
H13 |
109.144 |
| C3 |
C5 |
C7 |
111.021 |
|
C3 |
C5 |
H14 |
108.107 |
| C3 |
C5 |
H15 |
109.144 |
|
C4 |
C2 |
H8 |
110.309 |
| C4 |
C2 |
H9 |
110.911 |
|
C4 |
C6 |
H16 |
111.126 |
| C4 |
C6 |
H17 |
109.188 |
|
C4 |
C6 |
H18 |
110.554 |
| C5 |
C3 |
H10 |
110.309 |
|
C5 |
C3 |
H11 |
110.911 |
| C5 |
C7 |
H19 |
111.126 |
|
C5 |
C7 |
H20 |
109.188 |
| C5 |
C7 |
H21 |
110.554 |
|
C6 |
C4 |
H12 |
109.752 |
| C6 |
C4 |
H13 |
110.580 |
|
C7 |
C5 |
H14 |
109.752 |
| C7 |
C5 |
H15 |
110.580 |
|
H8 |
C2 |
H9 |
108.184 |
| H10 |
C3 |
H11 |
108.184 |
|
H12 |
C4 |
H13 |
108.157 |
| H14 |
C5 |
H15 |
108.157 |
|
H16 |
C6 |
H17 |
109.135 |
| H16 |
C6 |
H18 |
108.226 |
|
H17 |
C6 |
H18 |
108.560 |
| H19 |
C7 |
H20 |
109.135 |
|
H19 |
C7 |
H21 |
108.226 |
| H20 |
C7 |
H21 |
108.560 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.656 |
|
|
|
| 2 |
C |
-0.039 |
|
|
|
| 3 |
C |
-0.039 |
|
|
|
| 4 |
C |
-0.457 |
|
|
|
| 5 |
C |
-0.457 |
|
|
|
| 6 |
C |
-0.585 |
|
|
|
| 7 |
C |
-0.585 |
|
|
|
| 8 |
H |
0.182 |
|
|
|
| 9 |
H |
0.187 |
|
|
|
| 10 |
H |
0.182 |
|
|
|
| 11 |
H |
0.187 |
|
|
|
| 12 |
H |
0.224 |
|
|
|
| 13 |
H |
0.200 |
|
|
|
| 14 |
H |
0.224 |
|
|
|
| 15 |
H |
0.200 |
|
|
|
| 16 |
H |
0.194 |
|
|
|
| 17 |
H |
0.233 |
|
|
|
| 18 |
H |
0.189 |
|
|
|
| 19 |
H |
0.194 |
|
|
|
| 20 |
H |
0.233 |
|
|
|
| 21 |
H |
0.189 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.459 |
1.459 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.403 |
0.518 |
0.000 |
| y |
0.518 |
-44.675 |
0.000 |
| z |
0.000 |
0.000 |
-45.493 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.319 |
0.518 |
0.000 |
| y |
0.518 |
1.773 |
0.000 |
| z |
0.000 |
0.000 |
0.546 |
|
| Polar |
|---|
| 3z2-r2 | 1.092 |
|---|
| x2-y2 | -2.728 |
|---|
| xy | 0.518 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.662 |
-0.426 |
0.000 |
| y |
-0.426 |
10.498 |
0.000 |
| z |
0.000 |
0.000 |
8.764 |
<r2> (average value of r
2) Å
2
| <r2> |
335.143 |
| (<r2>)1/2 |
18.307 |