Jump to
S1C2
Energy calculated at HF/6-311G**
| | hartrees |
|---|
| Energy at 0K | -997.101668 |
| Energy at 298.15K | -997.106526 |
| Nuclear repulsion energy | 194.553671 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3264 |
2965 |
0.00 |
|
|
|
| 2 |
Ag |
1617 |
1470 |
0.00 |
|
|
|
| 3 |
Ag |
1478 |
1343 |
0.00 |
|
|
|
| 4 |
Ag |
1134 |
1030 |
0.00 |
|
|
|
| 5 |
Ag |
827 |
751 |
0.00 |
|
|
|
| 6 |
Ag |
322 |
292 |
0.00 |
|
|
|
| 7 |
Au |
3344 |
3038 |
8.23 |
|
|
|
| 8 |
Au |
1254 |
1139 |
2.69 |
|
|
|
| 9 |
Au |
833 |
757 |
2.45 |
|
|
|
| 10 |
Au |
127 |
115 |
9.09 |
|
|
|
| 11 |
Bg |
3321 |
3017 |
0.00 |
|
|
|
| 12 |
Bg |
1410 |
1281 |
0.00 |
|
|
|
| 13 |
Bg |
1106 |
1004 |
0.00 |
|
|
|
| 14 |
Bu |
3269 |
2970 |
24.00 |
|
|
|
| 15 |
Bu |
1612 |
1465 |
5.84 |
|
|
|
| 16 |
Bu |
1383 |
1256 |
64.25 |
|
|
|
| 17 |
Bu |
769 |
699 |
147.82 |
|
|
|
| 18 |
Bu |
233 |
212 |
11.37 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13651.3 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 12402.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G**
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.474 |
0.590 |
0.000 |
| C2 |
-0.474 |
-0.590 |
0.000 |
| Cl3 |
-0.474 |
2.115 |
0.000 |
| Cl4 |
0.474 |
-2.115 |
0.000 |
| H5 |
1.093 |
0.601 |
0.882 |
| H6 |
1.093 |
0.601 |
-0.882 |
| H7 |
-1.093 |
-0.601 |
0.882 |
| H8 |
-1.093 |
-0.601 |
-0.882 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
Cl3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.5137 | 1.7958 | 2.7051 | 1.0776 | 1.0776 | 2.1566 | 2.1566 |
C2 | 1.5137 | | 2.7051 | 1.7958 | 2.1566 | 2.1566 | 1.0776 | 1.0776 | Cl3 | 1.7958 | 2.7051 | | 4.3351 | 2.3507 | 2.3507 | 2.9218 | 2.9218 | Cl4 | 2.7051 | 1.7958 | 4.3351 | | 2.9218 | 2.9218 | 2.3507 | 2.3507 | H5 | 1.0776 | 2.1566 | 2.3507 | 2.9218 | | 1.7648 | 2.4936 | 3.0550 | H6 | 1.0776 | 2.1566 | 2.3507 | 2.9218 | 1.7648 | | 3.0550 | 2.4936 | H7 | 2.1566 | 1.0776 | 2.9218 | 2.3507 | 2.4936 | 3.0550 | | 1.7648 | H8 | 2.1566 | 1.0776 | 2.9218 | 2.3507 | 3.0550 | 2.4936 | 1.7648 | |
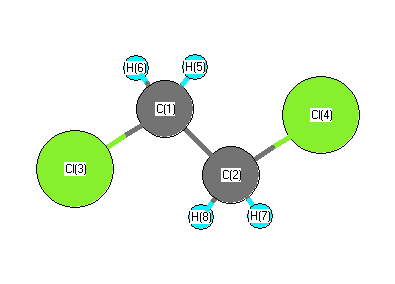 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
Cl4 |
109.347 |
|
C1 |
C2 |
H7 |
111.547 |
| C1 |
C2 |
H8 |
111.547 |
|
C2 |
C1 |
Cl3 |
109.347 |
| C2 |
C1 |
H5 |
111.547 |
|
C2 |
C1 |
H6 |
111.547 |
| Cl3 |
C1 |
H5 |
107.130 |
|
Cl3 |
C1 |
H6 |
107.130 |
| Cl4 |
C2 |
H7 |
107.130 |
|
Cl4 |
C2 |
H8 |
107.130 |
| H5 |
C1 |
H6 |
109.941 |
|
H7 |
C2 |
H8 |
109.941 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.196 |
|
|
|
| 2 |
C |
-0.196 |
|
|
|
| 3 |
Cl |
-0.141 |
|
|
|
| 4 |
Cl |
-0.141 |
|
|
|
| 5 |
H |
0.168 |
|
|
|
| 6 |
H |
0.168 |
|
|
|
| 7 |
H |
0.168 |
|
|
|
| 8 |
H |
0.168 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.031 |
2.499 |
0.000 |
| y |
2.499 |
-47.547 |
0.000 |
| z |
0.000 |
0.000 |
-38.313 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.899 |
2.499 |
0.000 |
| y |
2.499 |
-9.375 |
0.000 |
| z |
0.000 |
0.000 |
4.476 |
|
| Polar |
|---|
| 3z2-r2 | 8.952 |
|---|
| x2-y2 | 9.516 |
|---|
| xy | 2.499 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.325 |
-1.689 |
0.000 |
| y |
-1.689 |
8.802 |
0.000 |
| z |
0.000 |
0.000 |
4.151 |
<r2> (average value of r
2) Å
2
| <r2> |
201.739 |
| (<r2>)1/2 |
14.203 |
Jump to
S1C1
Energy calculated at HF/6-311G**
| | hartrees |
|---|
| Energy at 0K | -997.098817 |
| Energy at 298.15K | -997.103819 |
| HF Energy | -997.098817 |
| Nuclear repulsion energy | 201.802679 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3307 |
3005 |
1.77 |
|
|
|
| 2 |
A |
3252 |
2954 |
33.86 |
|
|
|
| 3 |
A |
1601 |
1455 |
0.25 |
|
|
|
| 4 |
A |
1479 |
1343 |
29.77 |
|
|
|
| 5 |
A |
1345 |
1222 |
1.49 |
|
|
|
| 6 |
A |
1131 |
1028 |
0.32 |
|
|
|
| 7 |
A |
1034 |
939 |
15.85 |
|
|
|
| 8 |
A |
708 |
643 |
33.21 |
|
|
|
| 9 |
A |
280 |
255 |
0.99 |
|
|
|
| 10 |
A |
123 |
112 |
1.27 |
|
|
|
| 11 |
B |
3320 |
3016 |
9.27 |
|
|
|
| 12 |
B |
3240 |
2944 |
4.88 |
|
|
|
| 13 |
B |
1593 |
1448 |
7.67 |
|
|
|
| 14 |
B |
1449 |
1316 |
59.08 |
|
|
|
| 15 |
B |
1270 |
1154 |
2.29 |
|
|
|
| 16 |
B |
977 |
888 |
22.74 |
|
|
|
| 17 |
B |
736 |
669 |
46.04 |
|
|
|
| 18 |
B |
439 |
399 |
9.66 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13642.7 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 12394.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.299 |
0.694 |
0.884 |
| C2 |
-0.299 |
-0.694 |
0.884 |
| Cl3 |
-0.299 |
1.707 |
-0.465 |
| Cl4 |
0.299 |
-1.707 |
-0.465 |
| H5 |
0.018 |
1.200 |
1.796 |
| H6 |
1.374 |
0.662 |
0.804 |
| H7 |
-0.018 |
-1.200 |
1.796 |
| H8 |
-1.374 |
-0.662 |
0.804 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
Cl3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.5119 | 1.7897 | 2.7541 | 1.0804 | 1.0781 | 2.1262 | 2.1550 |
C2 | 1.5119 | | 2.7541 | 1.7897 | 2.1262 | 2.1550 | 1.0804 | 1.0781 | Cl3 | 1.7897 | 2.7541 | | 3.4660 | 2.3386 | 2.3457 | 3.6936 | 2.8940 | Cl4 | 2.7541 | 1.7897 | 3.4660 | | 3.6936 | 2.8940 | 2.3386 | 2.3457 | H5 | 1.0804 | 2.1262 | 2.3386 | 3.6936 | | 1.7646 | 2.4005 | 2.5272 | H6 | 1.0781 | 2.1550 | 2.3457 | 2.8940 | 1.7646 | | 2.5272 | 3.0498 | H7 | 2.1262 | 1.0804 | 3.6936 | 2.3386 | 2.4005 | 2.5272 | | 1.7646 | H8 | 2.1550 | 1.0781 | 2.8940 | 2.3457 | 2.5272 | 3.0498 | 1.7646 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
Cl4 |
112.783 |
|
C1 |
C2 |
H7 |
109.073 |
| C1 |
C2 |
H8 |
111.521 |
|
C2 |
C1 |
Cl3 |
112.783 |
| C2 |
C1 |
H5 |
109.073 |
|
C2 |
C1 |
H6 |
111.521 |
| Cl3 |
C1 |
H5 |
106.505 |
|
Cl3 |
C1 |
H6 |
107.131 |
| Cl4 |
C2 |
H7 |
106.505 |
|
Cl4 |
C2 |
H8 |
107.131 |
| H5 |
C1 |
H6 |
109.674 |
|
H7 |
C2 |
H8 |
109.674 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.196 |
|
|
|
| 2 |
C |
-0.196 |
|
|
|
| 3 |
Cl |
-0.126 |
|
|
|
| 4 |
Cl |
-0.126 |
|
|
|
| 5 |
H |
0.152 |
|
|
|
| 6 |
H |
0.169 |
|
|
|
| 7 |
H |
0.152 |
|
|
|
| 8 |
H |
0.169 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.210 |
3.210 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.187 |
1.452 |
0.000 |
| y |
1.452 |
-44.110 |
0.000 |
| z |
0.000 |
0.000 |
-36.048 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.892 |
1.452 |
0.000 |
| y |
1.452 |
-6.992 |
0.000 |
| z |
0.000 |
0.000 |
5.100 |
|
| Polar |
|---|
| 3z2-r2 | 10.201 |
|---|
| x2-y2 | 5.923 |
|---|
| xy | 1.452 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.631 |
-0.682 |
0.000 |
| y |
-0.682 |
6.692 |
0.000 |
| z |
0.000 |
0.000 |
6.432 |
<r2> (average value of r
2) Å
2
| <r2> |
165.609 |
| (<r2>)1/2 |
12.869 |