Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3899 |
3542 |
129.36 |
|
|
|
| 2 |
A' |
3396 |
3085 |
2.66 |
|
|
|
| 3 |
A' |
3354 |
3047 |
21.08 |
|
|
|
| 4 |
A' |
3343 |
3037 |
17.27 |
|
|
|
| 5 |
A' |
1825 |
1658 |
119.89 |
|
|
|
| 6 |
A' |
1792 |
1628 |
167.74 |
|
|
|
| 7 |
A' |
1707 |
1551 |
42.70 |
|
|
|
| 8 |
A' |
1620 |
1471 |
10.84 |
|
|
|
| 9 |
A' |
1550 |
1408 |
87.57 |
|
|
|
| 10 |
A' |
1549 |
1408 |
56.96 |
|
|
|
| 11 |
A' |
1486 |
1350 |
72.72 |
|
|
|
| 12 |
A' |
1419 |
1289 |
25.19 |
|
|
|
| 13 |
A' |
1397 |
1270 |
7.59 |
|
|
|
| 14 |
A' |
1305 |
1185 |
6.00 |
|
|
|
| 15 |
A' |
1258 |
1143 |
45.65 |
|
|
|
| 16 |
A' |
1222 |
1110 |
12.11 |
|
|
|
| 17 |
A' |
1157 |
1051 |
29.44 |
|
|
|
| 18 |
A' |
1020 |
927 |
1.21 |
|
|
|
| 19 |
A' |
987 |
897 |
18.66 |
|
|
|
| 20 |
A' |
869 |
789 |
22.12 |
|
|
|
| 21 |
A' |
713 |
648 |
0.08 |
|
|
|
| 22 |
A' |
615 |
558 |
3.16 |
|
|
|
| 23 |
A' |
480 |
437 |
13.31 |
|
|
|
| 24 |
A" |
1131 |
1028 |
0.37 |
|
|
|
| 25 |
A" |
1070 |
973 |
5.58 |
|
|
|
| 26 |
A" |
1028 |
934 |
3.36 |
|
|
|
| 27 |
A" |
901 |
818 |
17.22 |
|
|
|
| 28 |
A" |
720 |
654 |
18.93 |
|
|
|
| 29 |
A" |
671 |
610 |
18.74 |
|
|
|
| 30 |
A" |
552 |
501 |
112.50 |
|
|
|
| 31 |
A" |
463 |
421 |
17.33 |
|
|
|
| 32 |
A" |
277 |
252 |
0.11 |
|
|
|
| 33 |
A" |
254 |
230 |
7.48 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22513.3 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 20453.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
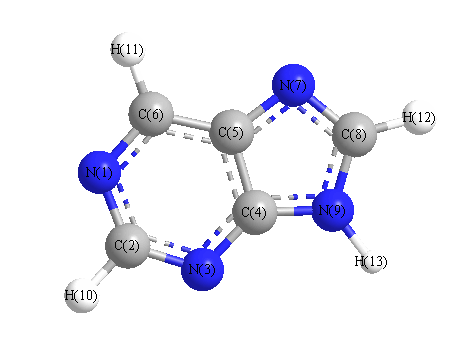
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.421 |
|
|
|
| 2 |
C |
0.231 |
|
|
|
| 3 |
N |
-0.452 |
|
|
|
| 4 |
C |
0.528 |
|
|
|
| 5 |
C |
-0.078 |
|
|
|
| 6 |
C |
0.224 |
|
|
|
| 7 |
N |
-0.425 |
|
|
|
| 8 |
C |
0.275 |
|
|
|
| 9 |
N |
-0.506 |
|
|
|
| 10 |
H |
0.117 |
|
|
|
| 11 |
H |
0.129 |
|
|
|
| 12 |
H |
0.123 |
|
|
|
| 13 |
H |
0.254 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.247 |
2.898 |
0.000 |
3.667 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-50.230 |
6.359 |
0.000 |
| y |
6.359 |
-47.653 |
0.000 |
| z |
0.000 |
0.000 |
-52.276 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.266 |
6.359 |
0.000 |
| y |
6.359 |
3.600 |
0.000 |
| z |
0.000 |
0.000 |
-3.335 |
|
| Polar |
|---|
| 3z2-r2 | -6.669 |
|---|
| x2-y2 | -2.577 |
|---|
| xy | 6.359 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.507 |
1.360 |
0.000 |
| y |
1.360 |
11.070 |
0.000 |
| z |
0.000 |
0.000 |
4.961 |
<r2> (average value of r
2) Å
2
| <r2> |
249.800 |
| (<r2>)1/2 |
15.805 |