Jump to
S1C2
Vibrational Frequencies calculated at HF/6-311G**
Geometric Data calculated at HF/6-311G**
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at HF/6-311G**
| | hartrees |
|---|
| Energy at 0K | -192.236399 |
| Energy at 298.15K | |
| HF Energy | -192.236399 |
| Nuclear repulsion energy | 149.278349 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3389 |
3079 |
0.37 |
|
|
|
| 2 |
A1 |
3379 |
3070 |
20.85 |
|
|
|
| 3 |
A1 |
3353 |
3046 |
0.66 |
|
|
|
| 4 |
A1 |
1720 |
1562 |
0.07 |
|
|
|
| 5 |
A1 |
1548 |
1407 |
0.14 |
|
|
|
| 6 |
A1 |
1217 |
1106 |
0.56 |
|
|
|
| 7 |
A1 |
1127 |
1024 |
8.11 |
|
|
|
| 8 |
A1 |
1078 |
979 |
3.26 |
|
|
|
| 9 |
A1 |
895 |
813 |
0.33 |
|
|
|
| 10 |
A2 |
1044 |
948 |
0.00 |
|
|
|
| 11 |
A2 |
821 |
746 |
0.00 |
|
|
|
| 12 |
A2 |
562 |
511 |
0.00 |
|
|
|
| 13 |
B1 |
921 |
836 |
15.03 |
|
|
|
| 14 |
B1 |
801 |
728 |
20.20 |
|
|
|
| 15 |
B1 |
710 |
645 |
100.12 |
|
|
|
| 16 |
B1 |
522 |
474 |
8.35 |
|
|
|
| 17 |
B2 |
3364 |
3056 |
54.92 |
|
|
|
| 18 |
B2 |
3353 |
3046 |
8.87 |
|
|
|
| 19 |
B2 |
1497 |
1360 |
47.14 |
|
|
|
| 20 |
B2 |
1400 |
1272 |
0.03 |
|
|
|
| 21 |
B2 |
1254 |
1139 |
14.39 |
|
|
|
| 22 |
B2 |
1014 |
921 |
1.48 |
|
|
|
| 23 |
B2 |
918 |
834 |
0.02 |
|
|
|
| 24 |
B2 |
3613i |
3282i |
12188.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16136.9 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 14660.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
1.222 |
| C2 |
0.000 |
1.125 |
0.395 |
| C3 |
0.000 |
0.668 |
-1.005 |
| C4 |
0.000 |
-0.668 |
-1.005 |
| C5 |
0.000 |
-1.125 |
0.395 |
| H6 |
0.000 |
0.000 |
2.294 |
| H7 |
0.000 |
2.150 |
0.711 |
| H8 |
0.000 |
1.314 |
-1.861 |
| H9 |
0.000 |
-1.314 |
-1.861 |
| H10 |
0.000 |
-2.150 |
0.711 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.3963 | 2.3250 | 2.3250 | 1.3963 | 1.0716 | 2.2104 | 3.3515 | 3.3515 | 2.2104 |
C2 | 1.3963 | | 1.4723 | 2.2744 | 2.2495 | 2.2070 | 1.0733 | 2.2637 | 3.3221 | 3.2904 | C3 | 2.3250 | 1.4723 | | 1.3359 | 2.2744 | 3.3656 | 2.2675 | 1.0726 | 2.1590 | 3.2996 | C4 | 2.3250 | 2.2744 | 1.3359 | | 1.4723 | 3.3656 | 3.2996 | 2.1590 | 1.0726 | 2.2675 | C5 | 1.3963 | 2.2495 | 2.2744 | 1.4723 | | 2.2070 | 3.2904 | 3.3221 | 2.2637 | 1.0733 | H6 | 1.0716 | 2.2070 | 3.3656 | 3.3656 | 2.2070 | | 2.6702 | 4.3576 | 4.3576 | 2.6702 | H7 | 2.2104 | 1.0733 | 2.2675 | 3.2996 | 3.2904 | 2.6702 | | 2.7045 | 4.3148 | 4.3009 | H8 | 3.3515 | 2.2637 | 1.0726 | 2.1590 | 3.3221 | 4.3576 | 2.7045 | | 2.6281 | 4.3148 | H9 | 3.3515 | 3.3221 | 2.1590 | 1.0726 | 2.2637 | 4.3576 | 4.3148 | 2.6281 | | 2.7045 | H10 | 2.2104 | 3.2904 | 3.2996 | 2.2675 | 1.0733 | 2.6702 | 4.3009 | 4.3148 | 2.7045 | |
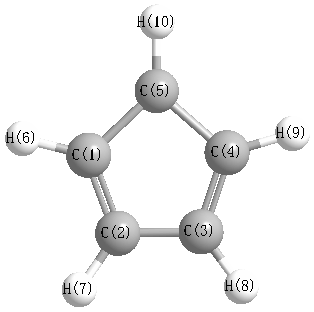 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
108.263 |
|
C1 |
C2 |
H7 |
126.535 |
| C1 |
C5 |
C4 |
108.263 |
|
C1 |
C5 |
H10 |
126.535 |
| C2 |
C1 |
C5 |
107.326 |
|
C2 |
C1 |
H6 |
126.337 |
| C2 |
C3 |
C4 |
108.074 |
|
C2 |
C3 |
H8 |
124.886 |
| C3 |
C2 |
H7 |
125.202 |
|
C3 |
C4 |
C5 |
108.074 |
| C3 |
C4 |
H9 |
127.040 |
|
C4 |
C3 |
H8 |
127.040 |
| C4 |
C5 |
H10 |
125.202 |
|
C5 |
C1 |
H6 |
126.337 |
| C5 |
C4 |
H9 |
124.886 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.166 |
|
|
|
| 2 |
C |
-0.058 |
|
|
|
| 3 |
C |
-0.120 |
|
|
|
| 4 |
C |
-0.120 |
|
|
|
| 5 |
C |
-0.058 |
|
|
|
| 6 |
H |
0.098 |
|
|
|
| 7 |
H |
0.110 |
|
|
|
| 8 |
H |
0.103 |
|
|
|
| 9 |
H |
0.103 |
|
|
|
| 10 |
H |
0.110 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.039 |
0.039 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.629 |
0.000 |
0.000 |
| y |
0.000 |
-25.639 |
0.000 |
| z |
0.000 |
0.000 |
-27.694 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.962 |
0.000 |
0.000 |
| y |
0.000 |
5.022 |
0.000 |
| z |
0.000 |
0.000 |
1.939 |
|
| Polar |
|---|
| 3z2-r2 | 3.879 |
|---|
| x2-y2 | -7.990 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.588 |
0.000 |
0.000 |
| y |
0.000 |
12.111 |
0.000 |
| z |
0.000 |
0.000 |
7.467 |
<r2> (average value of r
2) Å
2
| <r2> |
87.490 |
| (<r2>)1/2 |
9.354 |