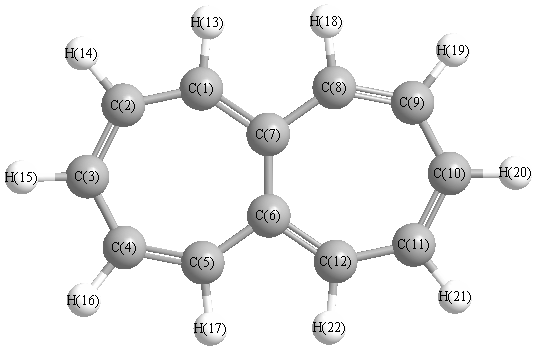
Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3327 |
3022 |
1.54 |
|
|
|
| 2 |
A |
3314 |
3011 |
38.81 |
|
|
|
| 3 |
A |
3303 |
3001 |
0.37 |
|
|
|
| 4 |
A |
3289 |
2988 |
0.72 |
|
|
|
| 5 |
A |
3287 |
2986 |
0.45 |
|
|
|
| 6 |
A |
1874 |
1702 |
5.78 |
|
|
|
| 7 |
A |
1815 |
1649 |
1.68 |
|
|
|
| 8 |
A |
1795 |
1631 |
0.03 |
|
|
|
| 9 |
A |
1588 |
1443 |
0.03 |
|
|
|
| 10 |
A |
1551 |
1409 |
0.39 |
|
|
|
| 11 |
A |
1522 |
1383 |
1.24 |
|
|
|
| 12 |
A |
1357 |
1233 |
0.00 |
|
|
|
| 13 |
A |
1344 |
1221 |
0.12 |
|
|
|
| 14 |
A |
1299 |
1181 |
0.28 |
|
|
|
| 15 |
A |
1122 |
1019 |
0.01 |
|
|
|
| 16 |
A |
1119 |
1017 |
1.21 |
|
|
|
| 17 |
A |
1102 |
1001 |
0.31 |
|
|
|
| 18 |
A |
1018 |
925 |
9.90 |
|
|
|
| 19 |
A |
1005 |
913 |
9.16 |
|
|
|
| 20 |
A |
959 |
871 |
4.24 |
|
|
|
| 21 |
A |
903 |
821 |
2.50 |
|
|
|
| 22 |
A |
853 |
775 |
2.13 |
|
|
|
| 23 |
A |
789 |
717 |
72.07 |
|
|
|
| 24 |
A |
702 |
638 |
2.48 |
|
|
|
| 25 |
A |
613 |
557 |
0.18 |
|
|
|
| 26 |
A |
469 |
426 |
7.58 |
|
|
|
| 27 |
A |
420 |
382 |
6.19 |
|
|
|
| 28 |
A |
378 |
343 |
0.33 |
|
|
|
| 29 |
A |
312 |
284 |
1.30 |
|
|
|
| 30 |
A |
142 |
129 |
1.29 |
|
|
|
| 31 |
A |
81 |
74 |
0.01 |
|
|
|
| 32 |
B |
3325 |
3021 |
101.28 |
|
|
|
| 33 |
B |
3316 |
3012 |
62.34 |
|
|
|
| 34 |
B |
3301 |
2999 |
15.15 |
|
|
|
| 35 |
B |
3292 |
2991 |
2.65 |
|
|
|
| 36 |
B |
3285 |
2985 |
14.36 |
|
|
|
| 37 |
B |
1837 |
1669 |
7.05 |
|
|
|
| 38 |
B |
1821 |
1654 |
16.33 |
|
|
|
| 39 |
B |
1744 |
1585 |
11.39 |
|
|
|
| 40 |
B |
1586 |
1441 |
2.01 |
|
|
|
| 41 |
B |
1525 |
1385 |
0.76 |
|
|
|
| 42 |
B |
1446 |
1314 |
0.71 |
|
|
|
| 43 |
B |
1355 |
1231 |
1.86 |
|
|
|
| 44 |
B |
1338 |
1216 |
0.50 |
|
|
|
| 45 |
B |
1186 |
1078 |
0.63 |
|
|
|
| 46 |
B |
1122 |
1020 |
0.17 |
|
|
|
| 47 |
B |
1115 |
1013 |
0.51 |
|
|
|
| 48 |
B |
1102 |
1001 |
1.14 |
|
|
|
| 49 |
B |
1029 |
935 |
1.43 |
|
|
|
| 50 |
B |
978 |
889 |
0.87 |
|
|
|
| 51 |
B |
950 |
863 |
17.30 |
|
|
|
| 52 |
B |
881 |
801 |
40.65 |
|
|
|
| 53 |
B |
830 |
754 |
25.47 |
|
|
|
| 54 |
B |
718 |
652 |
19.88 |
|
|
|
| 55 |
B |
652 |
593 |
8.61 |
|
|
|
| 56 |
B |
622 |
565 |
7.14 |
|
|
|
| 57 |
B |
451 |
410 |
1.79 |
|
|
|
| 58 |
B |
391 |
355 |
2.74 |
|
|
|
| 59 |
B |
297 |
270 |
0.23 |
|
|
|
| 60 |
B |
187 |
170 |
1.03 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 42167.9 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 38309.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.077 |
|
|
|
| 2 |
C |
-0.091 |
|
|
|
| 3 |
C |
-0.116 |
|
|
|
| 4 |
C |
-0.077 |
|
|
|
| 5 |
C |
-0.113 |
|
|
|
| 6 |
C |
-0.039 |
|
|
|
| 7 |
C |
-0.039 |
|
|
|
| 8 |
C |
-0.113 |
|
|
|
| 9 |
C |
-0.077 |
|
|
|
| 10 |
C |
-0.116 |
|
|
|
| 11 |
C |
-0.091 |
|
|
|
| 12 |
C |
-0.077 |
|
|
|
| 13 |
H |
0.093 |
|
|
|
| 14 |
H |
0.102 |
|
|
|
| 15 |
H |
0.102 |
|
|
|
| 16 |
H |
0.103 |
|
|
|
| 17 |
H |
0.114 |
|
|
|
| 18 |
H |
0.114 |
|
|
|
| 19 |
H |
0.103 |
|
|
|
| 20 |
H |
0.102 |
|
|
|
| 21 |
H |
0.102 |
|
|
|
| 22 |
H |
0.093 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.156 |
0.156 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-63.068 |
0.036 |
0.000 |
| y |
0.036 |
-65.556 |
0.000 |
| z |
0.000 |
0.000 |
-73.747 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.584 |
0.036 |
0.000 |
| y |
0.036 |
2.852 |
0.000 |
| z |
0.000 |
0.000 |
-9.435 |
|
| Polar |
|---|
| 3z2-r2 | -18.871 |
|---|
| x2-y2 | 2.488 |
|---|
| xy | 0.036 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
25.427 |
-3.604 |
0.000 |
| y |
-3.604 |
19.962 |
0.000 |
| z |
0.000 |
0.000 |
10.855 |
<r2> (average value of r
2) Å
2
| <r2> |
537.368 |
| (<r2>)1/2 |
23.181 |