Jump to
S1C2
Energy calculated at HF/6-311G**
| | hartrees |
|---|
| Energy at 0K | -191.958925 |
| Energy at 298.15K | -191.966418 |
| HF Energy | -191.958925 |
| Nuclear repulsion energy | 129.453424 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3216 |
2922 |
28.81 |
|
|
|
| 2 |
A1 |
3188 |
2896 |
11.24 |
|
|
|
| 3 |
A1 |
1677 |
1523 |
0.00 |
|
|
|
| 4 |
A1 |
1615 |
1467 |
4.66 |
|
|
|
| 5 |
A1 |
1522 |
1382 |
8.44 |
|
|
|
| 6 |
A1 |
1141 |
1036 |
24.91 |
|
|
|
| 7 |
A1 |
1024 |
930 |
32.34 |
|
|
|
| 8 |
A1 |
902 |
820 |
5.00 |
|
|
|
| 9 |
A2 |
3225 |
2930 |
0.00 |
|
|
|
| 10 |
A2 |
1338 |
1215 |
0.00 |
|
|
|
| 11 |
A2 |
1266 |
1151 |
0.00 |
|
|
|
| 12 |
A2 |
899 |
817 |
0.00 |
|
|
|
| 13 |
B1 |
3267 |
2968 |
86.39 |
|
|
|
| 14 |
B1 |
3222 |
2927 |
71.05 |
|
|
|
| 15 |
B1 |
1312 |
1192 |
0.02 |
|
|
|
| 16 |
B1 |
1265 |
1150 |
7.30 |
|
|
|
| 17 |
B1 |
824 |
748 |
0.03 |
|
|
|
| 18 |
B1 |
110 |
100 |
4.61 |
|
|
|
| 19 |
B2 |
3180 |
2889 |
173.45 |
|
|
|
| 20 |
B2 |
1646 |
1496 |
2.61 |
|
|
|
| 21 |
B2 |
1442 |
1310 |
0.01 |
|
|
|
| 22 |
B2 |
1388 |
1261 |
8.67 |
|
|
|
| 23 |
B2 |
1164 |
1058 |
131.25 |
|
|
|
| 24 |
B2 |
1002 |
911 |
1.46 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20416.9 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 18548.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
1.049 |
| C2 |
0.000 |
0.000 |
-1.072 |
| C3 |
0.000 |
1.027 |
0.072 |
| C4 |
0.000 |
-1.027 |
0.072 |
| H5 |
0.884 |
0.000 |
-1.697 |
| H6 |
-0.884 |
0.000 |
-1.697 |
| H7 |
0.884 |
1.652 |
0.143 |
| H8 |
-0.884 |
1.652 |
0.143 |
| H9 |
-0.884 |
-1.652 |
0.143 |
| H10 |
0.884 |
-1.652 |
0.143 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| O1 | | 2.1205 | 1.4181 | 1.4181 | 2.8842 | 2.8842 | 2.0811 | 2.0811 | 2.0811 | 2.0811 |
C2 | 2.1205 | | 1.5369 | 1.5369 | 1.0823 | 1.0823 | 2.2328 | 2.2328 | 2.2328 | 2.2328 | C3 | 1.4181 | 1.5369 | | 2.0548 | 2.2277 | 2.2277 | 1.0848 | 1.0848 | 2.8222 | 2.8222 | C4 | 1.4181 | 1.5369 | 2.0548 | | 2.2277 | 2.2277 | 2.8222 | 2.8222 | 1.0848 | 1.0848 | H5 | 2.8842 | 1.0823 | 2.2277 | 2.2277 | | 1.7671 | 2.4723 | 3.0393 | 3.0393 | 2.4723 | H6 | 2.8842 | 1.0823 | 2.2277 | 2.2277 | 1.7671 | | 3.0393 | 2.4723 | 2.4723 | 3.0393 | H7 | 2.0811 | 2.2328 | 1.0848 | 2.8222 | 2.4723 | 3.0393 | | 1.7684 | 3.7471 | 3.3035 | H8 | 2.0811 | 2.2328 | 1.0848 | 2.8222 | 3.0393 | 2.4723 | 1.7684 | | 3.3035 | 3.7471 | H9 | 2.0811 | 2.2328 | 2.8222 | 1.0848 | 3.0393 | 2.4723 | 3.7471 | 3.3035 | | 1.7684 | H10 | 2.0811 | 2.2328 | 2.8222 | 1.0848 | 2.4723 | 3.0393 | 3.3035 | 3.7471 | 1.7684 | |
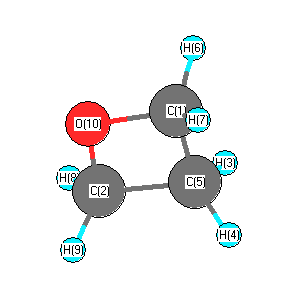 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C3 |
C2 |
91.622 |
|
O1 |
C3 |
H7 |
111.814 |
| O1 |
C3 |
H8 |
111.814 |
|
O1 |
C4 |
C2 |
91.622 |
| O1 |
C4 |
H9 |
111.814 |
|
O1 |
C4 |
H10 |
111.814 |
| C2 |
C3 |
H7 |
115.706 |
|
C2 |
C3 |
H8 |
115.706 |
| C2 |
C4 |
H9 |
115.706 |
|
C2 |
C4 |
H10 |
115.706 |
| C3 |
O1 |
C4 |
92.856 |
|
C3 |
C2 |
C4 |
83.899 |
| C3 |
C2 |
H5 |
115.436 |
|
C3 |
C2 |
H6 |
115.436 |
| C4 |
C2 |
H5 |
115.436 |
|
C4 |
C2 |
H6 |
115.436 |
| H5 |
C2 |
H6 |
109.451 |
|
H7 |
C3 |
H8 |
109.195 |
| H9 |
C4 |
H10 |
109.195 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.482 |
|
|
|
| 2 |
C |
-0.368 |
|
|
|
| 3 |
C |
0.160 |
|
|
|
| 4 |
C |
0.160 |
|
|
|
| 5 |
H |
0.112 |
|
|
|
| 6 |
H |
0.112 |
|
|
|
| 7 |
H |
0.076 |
|
|
|
| 8 |
H |
0.076 |
|
|
|
| 9 |
H |
0.076 |
|
|
|
| 10 |
H |
0.076 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-2.108 |
2.108 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.100 |
0.000 |
0.000 |
| y |
0.000 |
-22.751 |
0.000 |
| z |
0.000 |
0.000 |
-28.129 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.341 |
0.000 |
0.000 |
| y |
0.000 |
3.363 |
0.000 |
| z |
0.000 |
0.000 |
-4.704 |
|
| Polar |
|---|
| 3z2-r2 | -9.408 |
|---|
| x2-y2 | -1.348 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.591 |
0.000 |
0.000 |
| y |
0.000 |
5.511 |
0.000 |
| z |
0.000 |
0.000 |
4.615 |
<r2> (average value of r
2) Å
2
| <r2> |
65.471 |
| (<r2>)1/2 |
8.091 |
Jump to
S1C1
Energy calculated at HF/6-311G**
| | hartrees |
|---|
| Energy at 0K | -191.958925 |
| Energy at 298.15K | -191.966417 |
| HF Energy | -191.958925 |
| Nuclear repulsion energy | 129.451145 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3268 |
2969 |
85.88 |
|
|
|
| 2 |
A' |
3222 |
2927 |
71.55 |
|
|
|
| 3 |
A' |
3216 |
2922 |
28.89 |
|
|
|
| 4 |
A' |
3188 |
2896 |
11.14 |
|
|
|
| 5 |
A' |
1677 |
1523 |
0.00 |
|
|
|
| 6 |
A' |
1614 |
1467 |
4.66 |
|
|
|
| 7 |
A' |
1522 |
1382 |
8.45 |
|
|
|
| 8 |
A' |
1312 |
1192 |
0.02 |
|
|
|
| 9 |
A' |
1265 |
1150 |
7.30 |
|
|
|
| 10 |
A' |
1141 |
1036 |
25.01 |
|
|
|
| 11 |
A' |
1024 |
930 |
32.24 |
|
|
|
| 12 |
A' |
902 |
820 |
4.99 |
|
|
|
| 13 |
A' |
824 |
748 |
0.03 |
|
|
|
| 14 |
A' |
110 |
100 |
4.61 |
|
|
|
| 15 |
A" |
3225 |
2930 |
0.00 |
|
|
|
| 16 |
A" |
3180 |
2889 |
173.46 |
|
|
|
| 17 |
A" |
1646 |
1496 |
2.61 |
|
|
|
| 18 |
A" |
1442 |
1310 |
0.01 |
|
|
|
| 19 |
A" |
1387 |
1261 |
8.67 |
|
|
|
| 20 |
A" |
1338 |
1215 |
0.00 |
|
|
|
| 21 |
A" |
1266 |
1151 |
0.00 |
|
|
|
| 22 |
A" |
1165 |
1058 |
131.26 |
|
|
|
| 23 |
A" |
1002 |
910 |
1.45 |
|
|
|
| 24 |
A" |
899 |
817 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20416.6 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 18548.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
-0.001 |
-1.049 |
0.000 |
| C2 |
0.000 |
1.072 |
0.000 |
| C3 |
0.000 |
-0.072 |
1.027 |
| C4 |
0.000 |
-0.072 |
-1.027 |
| H5 |
0.884 |
1.697 |
0.000 |
| H6 |
-0.884 |
1.696 |
0.000 |
| H7 |
0.885 |
-0.144 |
1.651 |
| H8 |
-0.884 |
-0.143 |
1.652 |
| H9 |
0.885 |
-0.144 |
-1.651 |
| H10 |
-0.884 |
-0.143 |
-1.652 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| O1 | | 2.1205 | 1.4180 | 1.4180 | 2.8843 | 2.8839 | 2.0810 | 2.0811 | 2.0810 | 2.0811 |
C2 | 2.1205 | | 1.5371 | 1.5371 | 1.0822 | 1.0823 | 2.2330 | 2.2329 | 2.2330 | 2.2329 | C3 | 1.4180 | 1.5371 | | 2.0549 | 2.2277 | 2.2277 | 1.0848 | 1.0848 | 2.8221 | 2.8226 | C4 | 1.4180 | 1.5371 | 2.0549 | | 2.2277 | 2.2277 | 2.8221 | 2.8226 | 1.0848 | 1.0848 | H5 | 2.8843 | 1.0822 | 2.2277 | 2.2277 | | 1.7673 | 2.4725 | 3.0392 | 2.4725 | 3.0392 | H6 | 2.8839 | 1.0823 | 2.2277 | 2.2277 | 1.7673 | | 3.0396 | 2.4723 | 3.0396 | 2.4723 | H7 | 2.0810 | 2.2330 | 1.0848 | 2.8221 | 2.4725 | 3.0396 | | 1.7684 | 3.3030 | 3.7472 | H8 | 2.0811 | 2.2329 | 1.0848 | 2.8226 | 3.0392 | 2.4723 | 1.7684 | | 3.7472 | 3.3043 | H9 | 2.0810 | 2.2330 | 2.8221 | 1.0848 | 2.4725 | 3.0396 | 3.3030 | 3.7472 | | 1.7684 | H10 | 2.0811 | 2.2329 | 2.8226 | 1.0848 | 3.0392 | 2.4723 | 3.7472 | 3.3043 | 1.7684 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C3 |
C2 |
91.619 |
|
O1 |
C3 |
H7 |
111.809 |
| O1 |
C3 |
H8 |
111.822 |
|
O1 |
C4 |
C2 |
91.619 |
| O1 |
C4 |
H9 |
111.809 |
|
O1 |
C4 |
H10 |
111.822 |
| C2 |
C3 |
H7 |
115.710 |
|
C2 |
C3 |
H8 |
115.702 |
| C2 |
C4 |
H9 |
115.710 |
|
C2 |
C4 |
H10 |
115.702 |
| C3 |
O1 |
C4 |
92.867 |
|
C3 |
C2 |
C4 |
83.894 |
| C3 |
C2 |
H5 |
115.433 |
|
C3 |
C2 |
H6 |
115.431 |
| C4 |
C2 |
H5 |
115.433 |
|
C4 |
C2 |
H6 |
115.431 |
| H5 |
C2 |
H6 |
109.467 |
|
H7 |
C3 |
H8 |
109.194 |
| H9 |
C4 |
H10 |
109.194 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.482 |
|
|
|
| 2 |
C |
-0.368 |
|
|
|
| 3 |
C |
0.160 |
|
|
|
| 4 |
C |
0.160 |
|
|
|
| 5 |
H |
0.112 |
|
|
|
| 6 |
H |
0.112 |
|
|
|
| 7 |
H |
0.076 |
|
|
|
| 8 |
H |
0.076 |
|
|
|
| 9 |
H |
0.076 |
|
|
|
| 10 |
H |
0.076 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.001 |
2.107 |
0.000 |
2.107 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.100 |
-0.003 |
0.000 |
| y |
-0.003 |
-28.129 |
0.000 |
| z |
0.000 |
0.000 |
-22.751 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.341 |
-0.003 |
0.000 |
| y |
-0.003 |
-4.704 |
0.000 |
| z |
0.000 |
0.000 |
3.363 |
|
| Polar |
|---|
| 3z2-r2 | 6.727 |
|---|
| x2-y2 | 4.030 |
|---|
| xy | -0.003 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.591 |
-0.000 |
0.000 |
| y |
-0.000 |
4.615 |
0.000 |
| z |
0.000 |
0.000 |
5.512 |
<r2> (average value of r
2) Å
2
| <r2> |
65.474 |
| (<r2>)1/2 |
8.092 |