Jump to
S1C2
Energy calculated at HF/6-311G**
| | hartrees |
|---|
| Energy at 0K | -340.615384 |
| Energy at 298.15K | |
| HF Energy | -340.615384 |
| Nuclear repulsion energy | 249.407680 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3217 |
2923 |
66.66 |
|
|
|
| 2 |
A1 |
2111 |
1918 |
866.50 |
|
|
|
| 3 |
A1 |
1684 |
1530 |
4.61 |
|
|
|
| 4 |
A1 |
1539 |
1398 |
5.29 |
|
|
|
| 5 |
A1 |
1365 |
1240 |
322.05 |
|
|
|
| 6 |
A1 |
1019 |
926 |
17.92 |
|
|
|
| 7 |
A1 |
925 |
840 |
34.81 |
|
|
|
| 8 |
A1 |
790 |
718 |
1.96 |
|
|
|
| 9 |
A2 |
3236 |
2940 |
0.00 |
|
|
|
| 10 |
A2 |
1366 |
1241 |
0.00 |
|
|
|
| 11 |
A2 |
1276 |
1160 |
0.00 |
|
|
|
| 12 |
A2 |
191i |
173i |
0.00 |
|
|
|
| 13 |
B1 |
3262 |
2963 |
73.62 |
|
|
|
| 14 |
B1 |
1382 |
1256 |
4.31 |
|
|
|
| 15 |
B1 |
923 |
838 |
5.04 |
|
|
|
| 16 |
B1 |
875 |
795 |
46.71 |
|
|
|
| 17 |
B1 |
124 |
113 |
1.79 |
|
|
|
| 18 |
B2 |
3205 |
2912 |
36.09 |
|
|
|
| 19 |
B2 |
1672 |
1519 |
6.49 |
|
|
|
| 20 |
B2 |
1550 |
1408 |
32.51 |
|
|
|
| 21 |
B2 |
1336 |
1214 |
3.48 |
|
|
|
| 22 |
B2 |
1172 |
1065 |
367.15 |
|
|
|
| 23 |
B2 |
828 |
752 |
0.67 |
|
|
|
| 24 |
B2 |
564 |
513 |
0.37 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17615.7 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 16003.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.857 |
| O2 |
0.000 |
0.000 |
2.015 |
| O3 |
0.000 |
1.108 |
0.058 |
| O4 |
0.000 |
-1.108 |
0.058 |
| C5 |
0.000 |
0.772 |
-1.267 |
| C6 |
0.000 |
-0.772 |
-1.267 |
| H7 |
-0.879 |
1.181 |
-1.747 |
| H8 |
0.879 |
1.181 |
-1.747 |
| H9 |
0.879 |
-1.181 |
-1.747 |
| H10 |
-0.879 |
-1.181 |
-1.747 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1584 | 1.3662 | 1.3662 | 2.2596 | 2.2596 | 2.9918 | 2.9918 | 2.9918 | 2.9918 |
O2 | 1.1584 | | 2.2496 | 2.2496 | 3.3716 | 3.3716 | 4.0406 | 4.0406 | 4.0406 | 4.0406 | O3 | 1.3662 | 2.2496 | | 2.2158 | 1.3662 | 2.2992 | 2.0090 | 2.0090 | 3.0448 | 3.0448 | O4 | 1.3662 | 2.2496 | 2.2158 | | 2.2992 | 1.3662 | 3.0448 | 3.0448 | 2.0090 | 2.0090 | C5 | 2.2596 | 3.3716 | 1.3662 | 2.2992 | | 1.5434 | 1.0826 | 1.0826 | 2.1950 | 2.1950 | C6 | 2.2596 | 3.3716 | 2.2992 | 1.3662 | 1.5434 | | 2.1950 | 2.1950 | 1.0826 | 1.0826 | H7 | 2.9918 | 4.0406 | 2.0090 | 3.0448 | 1.0826 | 2.1950 | | 1.7589 | 2.9452 | 2.3623 | H8 | 2.9918 | 4.0406 | 2.0090 | 3.0448 | 1.0826 | 2.1950 | 1.7589 | | 2.3623 | 2.9452 | H9 | 2.9918 | 4.0406 | 3.0448 | 2.0090 | 2.1950 | 1.0826 | 2.9452 | 2.3623 | | 1.7589 | H10 | 2.9918 | 4.0406 | 3.0448 | 2.0090 | 2.1950 | 1.0826 | 2.3623 | 2.9452 | 1.7589 | |
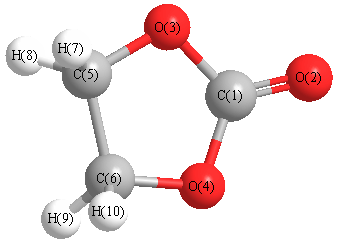 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
111.569 |
|
C1 |
O4 |
C6 |
111.569 |
| O2 |
C1 |
O3 |
125.815 |
|
O2 |
C1 |
O4 |
125.815 |
| O3 |
C1 |
O4 |
108.370 |
|
O3 |
C5 |
C6 |
104.246 |
| O3 |
C5 |
H7 |
109.707 |
|
O3 |
C5 |
H8 |
109.707 |
| O4 |
C6 |
C5 |
104.246 |
|
O4 |
C6 |
H9 |
109.707 |
| O4 |
C6 |
H10 |
109.707 |
|
C5 |
C6 |
H9 |
112.223 |
| C5 |
C6 |
H10 |
112.223 |
|
C6 |
C5 |
H7 |
112.223 |
| C6 |
C5 |
H8 |
112.223 |
|
H7 |
C5 |
H8 |
108.648 |
| H9 |
C6 |
H10 |
108.648 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.698 |
|
|
|
| 2 |
O |
-0.406 |
|
|
|
| 3 |
O |
-0.407 |
|
|
|
| 4 |
O |
-0.407 |
|
|
|
| 5 |
C |
0.044 |
|
|
|
| 6 |
C |
0.044 |
|
|
|
| 7 |
H |
0.108 |
|
|
|
| 8 |
H |
0.108 |
|
|
|
| 9 |
H |
0.108 |
|
|
|
| 10 |
H |
0.108 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.198 |
5.198 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.814 |
0.000 |
0.000 |
| y |
0.000 |
-36.759 |
0.000 |
| z |
0.000 |
0.000 |
-36.310 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.721 |
0.000 |
0.000 |
| y |
0.000 |
-2.697 |
0.000 |
| z |
0.000 |
0.000 |
-2.024 |
|
| Polar |
|---|
| 3z2-r2 | -4.048 |
|---|
| x2-y2 | 4.945 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.043 |
0.000 |
0.000 |
| y |
0.000 |
4.732 |
0.000 |
| z |
0.000 |
0.000 |
6.565 |
<r2> (average value of r
2) Å
2
| <r2> |
125.716 |
| (<r2>)1/2 |
11.212 |
Jump to
S1C1
Energy calculated at HF/6-311G**
| | hartrees |
|---|
| Energy at 0K | -340.615906 |
| Energy at 298.15K | -340.622467 |
| HF Energy | -340.615906 |
| Nuclear repulsion energy | 249.867563 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3260 |
2962 |
27.11 |
|
|
|
| 2 |
A |
3194 |
2902 |
32.29 |
|
|
|
| 3 |
A |
2114 |
1920 |
851.39 |
|
|
|
| 4 |
A |
1676 |
1523 |
5.27 |
|
|
|
| 5 |
A |
1545 |
1404 |
12.09 |
|
|
|
| 6 |
A |
1371 |
1246 |
39.93 |
|
|
|
| 7 |
A |
1358 |
1234 |
248.10 |
|
|
|
| 8 |
A |
1268 |
1152 |
22.80 |
|
|
|
| 9 |
A |
1017 |
924 |
8.85 |
|
|
|
| 10 |
A |
924 |
840 |
39.34 |
|
|
|
| 11 |
A |
788 |
716 |
2.21 |
|
|
|
| 12 |
A |
96 |
87 |
0.48 |
|
|
|
| 13 |
B |
3272 |
2973 |
50.71 |
|
|
|
| 14 |
B |
3195 |
2903 |
61.53 |
|
|
|
| 15 |
B |
1667 |
1514 |
7.54 |
|
|
|
| 16 |
B |
1543 |
1402 |
30.68 |
|
|
|
| 17 |
B |
1388 |
1261 |
10.05 |
|
|
|
| 18 |
B |
1316 |
1196 |
5.04 |
|
|
|
| 19 |
B |
1169 |
1062 |
349.38 |
|
|
|
| 20 |
B |
971 |
882 |
7.09 |
|
|
|
| 21 |
B |
881 |
801 |
49.16 |
|
|
|
| 22 |
B |
764 |
694 |
1.06 |
|
|
|
| 23 |
B |
565 |
513 |
0.29 |
|
|
|
| 24 |
B |
136 |
123 |
1.67 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17738.9 cm
-1
Scaled (by 0.9085) Zero Point Vibrational Energy (zpe) 16115.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.853 |
| O2 |
0.000 |
0.000 |
2.011 |
| O3 |
0.000 |
1.108 |
0.050 |
| O4 |
0.000 |
-1.108 |
0.050 |
| C5 |
0.171 |
0.747 |
-1.259 |
| C6 |
-0.171 |
-0.747 |
-1.259 |
| H7 |
-0.485 |
1.331 |
-1.889 |
| H8 |
1.198 |
0.926 |
-1.560 |
| H9 |
0.485 |
-1.331 |
-1.889 |
| H10 |
-1.198 |
-0.926 |
-1.560 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1583 | 1.3683 | 1.3683 | 2.2471 | 2.2471 | 3.0863 | 2.8487 | 3.0863 | 2.8487 |
O2 | 1.1583 | | 2.2528 | 2.2528 | 3.3592 | 3.3592 | 4.1495 | 3.8790 | 4.1495 | 3.8790 | O3 | 1.3683 | 2.2528 | | 2.2151 | 1.3683 | 2.2766 | 2.0107 | 2.0148 | 3.1527 | 2.8567 | O4 | 1.3683 | 2.2528 | 2.2151 | | 2.2766 | 1.3683 | 3.1527 | 2.8567 | 2.0107 | 2.0148 | C5 | 2.2471 | 3.3592 | 1.3683 | 2.2766 | | 1.5332 | 1.0807 | 1.0852 | 2.1942 | 2.1828 | C6 | 2.2471 | 3.3592 | 2.2766 | 1.3683 | 1.5332 | | 2.1942 | 2.1828 | 1.0807 | 1.0852 | H7 | 3.0863 | 4.1495 | 2.0107 | 3.1527 | 1.0807 | 2.1942 | | 1.7628 | 2.8335 | 2.3894 | H8 | 2.8487 | 3.8790 | 2.0148 | 2.8567 | 1.0852 | 2.1828 | 1.7628 | | 2.3894 | 3.0286 | H9 | 3.0863 | 4.1495 | 3.1527 | 2.0107 | 2.1942 | 1.0807 | 2.8335 | 2.3894 | | 1.7628 | H10 | 2.8487 | 3.8790 | 2.8567 | 2.0148 | 2.1828 | 1.0852 | 2.3894 | 3.0286 | 1.7628 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
110.401 |
|
C1 |
O4 |
C6 |
110.401 |
| O2 |
C1 |
O3 |
125.958 |
|
O2 |
C1 |
O4 |
125.958 |
| O3 |
C1 |
O4 |
108.084 |
|
O3 |
C5 |
C6 |
103.224 |
| O3 |
C5 |
H7 |
109.823 |
|
O3 |
C5 |
H8 |
109.880 |
| O4 |
C6 |
C5 |
103.224 |
|
O4 |
C6 |
H9 |
109.823 |
| O4 |
C6 |
H10 |
109.880 |
|
C5 |
C6 |
H9 |
113.016 |
| C5 |
C6 |
H10 |
111.807 |
|
C6 |
C5 |
H7 |
113.016 |
| C6 |
C5 |
H8 |
111.807 |
|
H7 |
C5 |
H8 |
108.955 |
| H9 |
C6 |
H10 |
108.955 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.699 |
|
|
|
| 2 |
O |
-0.403 |
|
|
|
| 3 |
O |
-0.407 |
|
|
|
| 4 |
O |
-0.407 |
|
|
|
| 5 |
C |
0.043 |
|
|
|
| 6 |
C |
0.043 |
|
|
|
| 7 |
H |
0.110 |
|
|
|
| 8 |
H |
0.106 |
|
|
|
| 9 |
H |
0.110 |
|
|
|
| 10 |
H |
0.106 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.158 |
5.158 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.885 |
0.204 |
0.000 |
| y |
0.204 |
-36.744 |
0.000 |
| z |
0.000 |
0.000 |
-36.151 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.563 |
0.204 |
0.000 |
| y |
0.204 |
-2.727 |
0.000 |
| z |
0.000 |
0.000 |
-1.837 |
|
| Polar |
|---|
| 3z2-r2 | -3.673 |
|---|
| x2-y2 | 4.860 |
|---|
| xy | 0.204 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.106 |
0.112 |
0.000 |
| y |
0.112 |
4.699 |
0.000 |
| z |
0.000 |
0.000 |
6.549 |
<r2> (average value of r
2) Å
2
| <r2> |
124.894 |
| (<r2>)1/2 |
11.176 |