Jump to
S1C2
Energy calculated at HF/TZVP
| | hartrees |
|---|
| Energy at 0K | -307.115432 |
| Energy at 298.15K | -307.127404 |
| HF Energy | -307.115432 |
| Nuclear repulsion energy | 246.354793 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
4169 |
3788 |
0.00 |
|
|
|
| 2 |
Ag |
3170 |
2881 |
0.00 |
|
|
|
| 3 |
Ag |
3139 |
2852 |
0.00 |
|
|
|
| 4 |
Ag |
1657 |
1505 |
0.00 |
|
|
|
| 5 |
Ag |
1621 |
1472 |
0.00 |
|
|
|
| 6 |
Ag |
1600 |
1454 |
0.00 |
|
|
|
| 7 |
Ag |
1515 |
1377 |
0.00 |
|
|
|
| 8 |
Ag |
1360 |
1236 |
0.00 |
|
|
|
| 9 |
Ag |
1174 |
1067 |
0.00 |
|
|
|
| 10 |
Ag |
1120 |
1017 |
0.00 |
|
|
|
| 11 |
Ag |
1111 |
1009 |
0.00 |
|
|
|
| 12 |
Ag |
387 |
352 |
0.00 |
|
|
|
| 13 |
Ag |
363 |
330 |
0.00 |
|
|
|
| 14 |
Au |
3225 |
2931 |
142.09 |
|
|
|
| 15 |
Au |
3160 |
2871 |
78.40 |
|
|
|
| 16 |
Au |
1442 |
1310 |
0.84 |
|
|
|
| 17 |
Au |
1332 |
1210 |
6.99 |
|
|
|
| 18 |
Au |
1023 |
929 |
2.91 |
|
|
|
| 19 |
Au |
812 |
737 |
0.07 |
|
|
|
| 20 |
Au |
290 |
263 |
279.36 |
|
|
|
| 21 |
Au |
102 |
93 |
20.98 |
|
|
|
| 22 |
Au |
88 |
80 |
6.38 |
|
|
|
| 23 |
Bg |
3197 |
2905 |
0.00 |
|
|
|
| 24 |
Bg |
3164 |
2875 |
0.00 |
|
|
|
| 25 |
Bg |
1429 |
1299 |
0.00 |
|
|
|
| 26 |
Bg |
1404 |
1276 |
0.00 |
|
|
|
| 27 |
Bg |
1284 |
1167 |
0.00 |
|
|
|
| 28 |
Bg |
881 |
801 |
0.00 |
|
|
|
| 29 |
Bg |
283 |
258 |
0.00 |
|
|
|
| 30 |
Bg |
169 |
153 |
0.00 |
|
|
|
| 31 |
Bu |
4169 |
3788 |
111.24 |
|
|
|
| 32 |
Bu |
3176 |
2886 |
101.20 |
|
|
|
| 33 |
Bu |
3139 |
2852 |
103.72 |
|
|
|
| 34 |
Bu |
1660 |
1508 |
8.50 |
|
|
|
| 35 |
Bu |
1629 |
1481 |
4.77 |
|
|
|
| 36 |
Bu |
1595 |
1449 |
19.98 |
|
|
|
| 37 |
Bu |
1421 |
1291 |
74.60 |
|
|
|
| 38 |
Bu |
1298 |
1180 |
106.27 |
|
|
|
| 39 |
Bu |
1178 |
1071 |
220.83 |
|
|
|
| 40 |
Bu |
1048 |
952 |
1.03 |
|
|
|
| 41 |
Bu |
560 |
509 |
42.55 |
|
|
|
| 42 |
Bu |
151 |
137 |
7.64 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 33346.5 cm
-1
Scaled (by 0.9086) Zero Point Vibrational Energy (zpe) 30298.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/TZVP
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
1.472 |
2.681 |
0.000 |
| O2 |
-1.472 |
-2.681 |
0.000 |
| C3 |
1.472 |
1.264 |
0.000 |
| C4 |
-1.472 |
-1.264 |
0.000 |
| C5 |
0.039 |
0.768 |
0.000 |
| C6 |
-0.039 |
-0.768 |
0.000 |
| H7 |
2.356 |
3.002 |
0.000 |
| H8 |
-2.356 |
-3.002 |
0.000 |
| H9 |
-0.456 |
1.168 |
0.877 |
| H10 |
-0.456 |
1.168 |
-0.877 |
| H11 |
0.456 |
-1.168 |
0.877 |
| H12 |
0.456 |
-1.168 |
-0.877 |
| H13 |
-1.983 |
-0.888 |
-0.882 |
| H14 |
-1.983 |
-0.888 |
0.882 |
| H15 |
1.983 |
0.888 |
-0.882 |
| H16 |
1.983 |
0.888 |
0.882 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| O1 | | 6.1168 | 1.4171 | 4.9220 | 2.3897 | 3.7656 | 0.9409 | 6.8517 | 2.6029 | 2.6029 | 4.0763 | 4.0763 | 5.0449 | 5.0449 | 2.0626 | 2.0626 |
O2 | 6.1168 | | 4.9220 | 1.4171 | 3.7656 | 2.3897 | 6.8517 | 0.9409 | 4.0763 | 4.0763 | 2.6029 | 2.6029 | 2.0626 | 2.0626 | 5.0449 | 5.0449 | C3 | 1.4171 | 4.9220 | | 3.8798 | 1.5156 | 2.5323 | 1.9499 | 5.7313 | 2.1203 | 2.1203 | 2.7776 | 2.7776 | 4.1646 | 4.1646 | 1.0865 | 1.0865 | C4 | 4.9220 | 1.4171 | 3.8798 | | 2.5323 | 1.5156 | 5.7313 | 1.9499 | 2.7776 | 2.7776 | 2.1203 | 2.1203 | 1.0865 | 1.0865 | 4.1646 | 4.1646 | C5 | 2.3897 | 3.7656 | 1.5156 | 2.5323 | | 1.5381 | 3.2181 | 4.4665 | 1.0841 | 1.0841 | 2.1661 | 2.1661 | 2.7588 | 2.7588 | 2.1377 | 2.1377 | C6 | 3.7656 | 2.3897 | 2.5323 | 1.5156 | 1.5381 | | 4.4665 | 3.2181 | 2.1661 | 2.1661 | 1.0841 | 1.0841 | 2.1377 | 2.1377 | 2.7588 | 2.7588 | H7 | 0.9409 | 6.8517 | 1.9499 | 5.7313 | 3.2181 | 4.4665 | | 7.6320 | 3.4701 | 3.4701 | 4.6654 | 4.6654 | 5.8938 | 5.8938 | 2.3204 | 2.3204 | H8 | 6.8517 | 0.9409 | 5.7313 | 1.9499 | 4.4665 | 3.2181 | 7.6320 | | 4.6654 | 4.6654 | 3.4701 | 3.4701 | 2.3204 | 2.3204 | 5.8938 | 5.8938 | H9 | 2.6029 | 4.0763 | 2.1203 | 2.7776 | 1.0841 | 2.1661 | 3.4701 | 4.6654 | | 1.7541 | 2.5083 | 3.0608 | 3.1068 | 2.5610 | 3.0205 | 2.4555 | H10 | 2.6029 | 4.0763 | 2.1203 | 2.7776 | 1.0841 | 2.1661 | 3.4701 | 4.6654 | 1.7541 | | 3.0608 | 2.5083 | 2.5610 | 3.1068 | 2.4555 | 3.0205 | H11 | 4.0763 | 2.6029 | 2.7776 | 2.1203 | 2.1661 | 1.0841 | 4.6654 | 3.4701 | 2.5083 | 3.0608 | | 1.7541 | 3.0205 | 2.4555 | 3.1068 | 2.5610 | H12 | 4.0763 | 2.6029 | 2.7776 | 2.1203 | 2.1661 | 1.0841 | 4.6654 | 3.4701 | 3.0608 | 2.5083 | 1.7541 | | 2.4555 | 3.0205 | 2.5610 | 3.1068 | H13 | 5.0449 | 2.0626 | 4.1646 | 1.0865 | 2.7588 | 2.1377 | 5.8938 | 2.3204 | 3.1068 | 2.5610 | 3.0205 | 2.4555 | | 1.7637 | 4.3457 | 4.6899 | H14 | 5.0449 | 2.0626 | 4.1646 | 1.0865 | 2.7588 | 2.1377 | 5.8938 | 2.3204 | 2.5610 | 3.1068 | 2.4555 | 3.0205 | 1.7637 | | 4.6899 | 4.3457 | H15 | 2.0626 | 5.0449 | 1.0865 | 4.1646 | 2.1377 | 2.7588 | 2.3204 | 5.8938 | 3.0205 | 2.4555 | 3.1068 | 2.5610 | 4.3457 | 4.6899 | | 1.7637 | H16 | 2.0626 | 5.0449 | 1.0865 | 4.1646 | 2.1377 | 2.7588 | 2.3204 | 5.8938 | 2.4555 | 3.0205 | 2.5610 | 3.1068 | 4.6899 | 4.3457 | 1.7637 | |
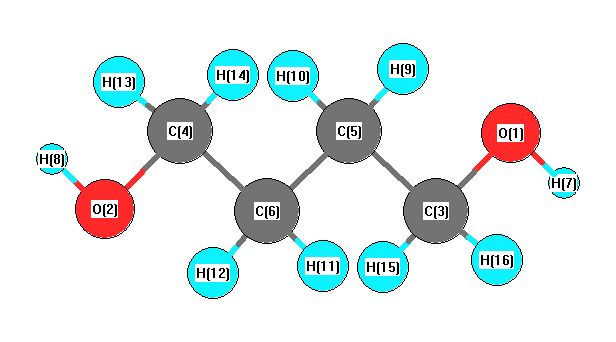 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C3 |
C5 |
109.094 |
|
O1 |
C3 |
H15 |
110.241 |
| O1 |
C3 |
H16 |
110.241 |
|
O2 |
C4 |
C6 |
109.094 |
| O2 |
C4 |
H13 |
110.241 |
|
O2 |
C4 |
H14 |
110.241 |
| C3 |
O1 |
H7 |
109.921 |
|
C3 |
C5 |
C6 |
112.038 |
| C3 |
C5 |
H9 |
108.149 |
|
C3 |
C5 |
H10 |
108.149 |
| C4 |
O2 |
H8 |
109.921 |
|
C4 |
C6 |
C5 |
112.038 |
| C4 |
C6 |
H11 |
108.149 |
|
C4 |
C6 |
H12 |
108.149 |
| C5 |
C3 |
H15 |
109.366 |
|
C5 |
C3 |
H16 |
109.366 |
| C5 |
C6 |
H11 |
110.190 |
|
C5 |
C6 |
H12 |
110.190 |
| C6 |
C4 |
H13 |
109.366 |
|
C6 |
C4 |
H14 |
109.366 |
| C6 |
C5 |
H9 |
110.190 |
|
C6 |
C5 |
H10 |
110.190 |
| H9 |
C5 |
H10 |
108.004 |
|
H11 |
C6 |
H12 |
108.004 |
| H13 |
C4 |
H14 |
108.514 |
|
H15 |
C3 |
H16 |
108.514 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.424 |
|
|
|
| 2 |
O |
-0.424 |
|
|
|
| 3 |
C |
-0.006 |
|
|
|
| 4 |
C |
-0.006 |
|
|
|
| 5 |
C |
-0.087 |
|
|
|
| 6 |
C |
-0.087 |
|
|
|
| 7 |
H |
0.272 |
|
|
|
| 8 |
H |
0.272 |
|
|
|
| 9 |
H |
0.069 |
|
|
|
| 10 |
H |
0.069 |
|
|
|
| 11 |
H |
0.069 |
|
|
|
| 12 |
H |
0.069 |
|
|
|
| 13 |
H |
0.053 |
|
|
|
| 14 |
H |
0.053 |
|
|
|
| 15 |
H |
0.053 |
|
|
|
| 16 |
H |
0.053 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-26.227 |
6.863 |
0.000 |
| y |
6.863 |
-46.471 |
0.000 |
| z |
0.000 |
0.000 |
-38.362 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
16.189 |
6.863 |
0.000 |
| y |
6.863 |
-14.176 |
0.000 |
| z |
0.000 |
0.000 |
-2.013 |
|
| Polar |
|---|
| 3z2-r2 | -4.027 |
|---|
| x2-y2 | 20.243 |
|---|
| xy | 6.863 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.006 |
0.900 |
0.000 |
| y |
0.900 |
8.410 |
0.000 |
| z |
0.000 |
0.000 |
6.794 |
<r2> (average value of r
2) Å
2
| <r2> |
283.258 |
| (<r2>)1/2 |
16.830 |
Jump to
S1C1
Energy calculated at HF/TZVP
| | hartrees |
|---|
| Energy at 0K | -307.114086 |
| Energy at 298.15K | |
| HF Energy | -307.114086 |
| Nuclear repulsion energy | 263.693789 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
4170 |
3789 |
58.59 |
|
|
|
| 2 |
A |
4148 |
3769 |
55.65 |
|
|
|
| 3 |
A |
3227 |
2932 |
81.03 |
|
|
|
| 4 |
A |
3217 |
2923 |
83.47 |
|
|
|
| 5 |
A |
3197 |
2905 |
6.03 |
|
|
|
| 6 |
A |
3192 |
2900 |
31.33 |
|
|
|
| 7 |
A |
3173 |
2883 |
32.60 |
|
|
|
| 8 |
A |
3165 |
2876 |
134.61 |
|
|
|
| 9 |
A |
3150 |
2862 |
7.79 |
|
|
|
| 10 |
A |
3146 |
2859 |
58.02 |
|
|
|
| 11 |
A |
1656 |
1505 |
3.21 |
|
|
|
| 12 |
A |
1649 |
1498 |
0.64 |
|
|
|
| 13 |
A |
1613 |
1465 |
8.63 |
|
|
|
| 14 |
A |
1602 |
1456 |
7.83 |
|
|
|
| 15 |
A |
1594 |
1448 |
1.94 |
|
|
|
| 16 |
A |
1570 |
1426 |
49.08 |
|
|
|
| 17 |
A |
1533 |
1393 |
1.71 |
|
|
|
| 18 |
A |
1510 |
1372 |
4.21 |
|
|
|
| 19 |
A |
1473 |
1338 |
15.88 |
|
|
|
| 20 |
A |
1434 |
1303 |
16.11 |
|
|
|
| 21 |
A |
1408 |
1280 |
10.71 |
|
|
|
| 22 |
A |
1367 |
1242 |
1.77 |
|
|
|
| 23 |
A |
1350 |
1227 |
58.55 |
|
|
|
| 24 |
A |
1245 |
1131 |
23.48 |
|
|
|
| 25 |
A |
1233 |
1120 |
7.33 |
|
|
|
| 26 |
A |
1200 |
1091 |
80.42 |
|
|
|
| 27 |
A |
1189 |
1080 |
43.31 |
|
|
|
| 28 |
A |
1121 |
1018 |
11.15 |
|
|
|
| 29 |
A |
1084 |
985 |
76.82 |
|
|
|
| 30 |
A |
1006 |
914 |
1.22 |
|
|
|
| 31 |
A |
956 |
868 |
5.08 |
|
|
|
| 32 |
A |
864 |
785 |
1.26 |
|
|
|
| 33 |
A |
838 |
762 |
0.95 |
|
|
|
| 34 |
A |
577 |
524 |
9.71 |
|
|
|
| 35 |
A |
481 |
437 |
9.03 |
|
|
|
| 36 |
A |
361 |
328 |
26.11 |
|
|
|
| 37 |
A |
357 |
324 |
7.64 |
|
|
|
| 38 |
A |
295 |
268 |
159.19 |
|
|
|
| 39 |
A |
262 |
238 |
71.89 |
|
|
|
| 40 |
A |
227 |
206 |
60.27 |
|
|
|
| 41 |
A |
117 |
106 |
10.33 |
|
|
|
| 42 |
A |
59 |
54 |
25.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 33507.1 cm
-1
Scaled (by 0.9086) Zero Point Vibrational Energy (zpe) 30444.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/TZVP
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-1.464 |
-0.004 |
0.500 |
| C2 |
-0.732 |
1.246 |
0.037 |
| C3 |
0.685 |
1.031 |
-0.510 |
| C4 |
1.588 |
0.133 |
0.328 |
| O5 |
-1.327 |
-1.039 |
-0.457 |
| O6 |
1.422 |
-1.232 |
0.034 |
| H7 |
-1.074 |
-0.337 |
1.458 |
| H8 |
-2.514 |
0.229 |
0.642 |
| H9 |
-0.697 |
1.924 |
0.886 |
| H10 |
-1.334 |
1.741 |
-0.719 |
| H11 |
0.638 |
0.616 |
-1.510 |
| H12 |
1.155 |
2.005 |
-0.610 |
| H13 |
1.398 |
0.321 |
1.385 |
| H14 |
2.626 |
0.382 |
0.152 |
| H15 |
-1.931 |
-1.740 |
-0.281 |
| H16 |
0.509 |
-1.409 |
-0.155 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5210 | 2.5915 | 3.0601 | 1.4166 | 3.1715 | 1.0863 | 1.0846 | 2.1111 | 2.1327 | 2.9743 | 3.4828 | 3.0137 | 4.1237 | 1.9598 | 2.5094 |
C2 | 1.5210 | | 1.5353 | 2.5898 | 2.4124 | 3.2840 | 2.1540 | 2.1388 | 1.0869 | 1.0854 | 2.1609 | 2.1347 | 2.6853 | 3.4700 | 3.2334 | 2.9370 | C3 | 2.5915 | 1.5353 | | 1.5247 | 2.8873 | 2.4417 | 2.9737 | 3.4943 | 2.1586 | 2.1504 | 1.0832 | 1.0855 | 2.1459 | 2.1509 | 3.8180 | 2.4717 | C4 | 3.0601 | 2.5898 | 1.5247 | | 3.2378 | 1.4061 | 2.9295 | 4.1149 | 2.9563 | 3.4953 | 2.1249 | 2.1384 | 1.0896 | 1.0826 | 4.0321 | 1.9426 | O5 | 1.4166 | 2.4124 | 2.8873 | 3.2378 | | 2.7991 | 2.0555 | 2.0561 | 3.3139 | 2.7922 | 2.7762 | 3.9302 | 3.5590 | 4.2443 | 0.9422 | 1.8966 | O6 | 3.1715 | 3.2840 | 2.4417 | 1.4061 | 2.7991 | | 3.0102 | 4.2427 | 3.8960 | 4.1233 | 2.5330 | 3.3113 | 2.0582 | 2.0169 | 3.4059 | 0.9493 | H7 | 1.0863 | 2.1540 | 2.9737 | 2.9295 | 2.0555 | 3.0102 | | 1.7489 | 2.3624 | 3.0201 | 3.5565 | 3.8378 | 2.5593 | 3.9896 | 2.3929 | 2.5018 | H8 | 1.0846 | 2.1388 | 3.4943 | 4.1149 | 2.0561 | 4.2427 | 1.7489 | | 2.4970 | 2.3518 | 3.8364 | 4.2642 | 3.9832 | 5.1661 | 2.2516 | 3.5296 | H9 | 2.1111 | 1.0869 | 2.1586 | 2.9563 | 3.3139 | 3.8960 | 2.3624 | 2.4970 | | 1.7362 | 3.0388 | 2.3821 | 2.6849 | 3.7367 | 4.0387 | 3.6942 | H10 | 2.1327 | 1.0854 | 2.1504 | 3.4953 | 2.7922 | 4.1233 | 3.0201 | 2.3518 | 1.7362 | | 2.4039 | 2.5049 | 3.7289 | 4.2762 | 3.5589 | 3.6922 | H11 | 2.9743 | 2.1609 | 1.0832 | 2.1249 | 2.7762 | 2.5330 | 3.5565 | 3.8364 | 3.0388 | 2.4039 | | 1.7336 | 3.0075 | 2.6017 | 3.6962 | 2.4395 | H12 | 3.4828 | 2.1347 | 1.0855 | 2.1384 | 3.9302 | 3.3113 | 3.8378 | 4.2642 | 2.3821 | 2.5049 | 1.7336 | | 2.6220 | 2.3194 | 4.8636 | 3.5037 | H13 | 3.0137 | 2.6853 | 2.1459 | 1.0896 | 3.5590 | 2.0582 | 2.5593 | 3.9832 | 2.6849 | 3.7289 | 3.0075 | 2.6220 | | 1.7416 | 4.2547 | 2.4806 | H14 | 4.1237 | 3.4700 | 2.1509 | 1.0826 | 4.2443 | 2.0169 | 3.9896 | 5.1661 | 3.7367 | 4.2762 | 2.6017 | 2.3194 | 1.7416 | | 5.0453 | 2.7897 | H15 | 1.9598 | 3.2334 | 3.8180 | 4.0321 | 0.9422 | 3.4059 | 2.3929 | 2.2516 | 4.0387 | 3.5589 | 3.6962 | 4.8636 | 4.2547 | 5.0453 | | 2.4654 | H16 | 2.5094 | 2.9370 | 2.4717 | 1.9426 | 1.8966 | 0.9493 | 2.5018 | 3.5296 | 3.6942 | 3.6922 | 2.4395 | 3.5037 | 2.4806 | 2.7897 | 2.4654 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C3 |
C5 |
29.338 |
|
O1 |
C3 |
H15 |
28.123 |
| O1 |
C3 |
H16 |
59.364 |
|
O2 |
C4 |
C6 |
106.763 |
| O2 |
C4 |
H13 |
83.047 |
|
O2 |
C4 |
H14 |
137.925 |
| C3 |
O1 |
H7 |
99.678 |
|
C3 |
C5 |
C6 |
50.829 |
| C3 |
C5 |
H9 |
40.002 |
|
C3 |
C5 |
H10 |
44.458 |
| C4 |
O2 |
H8 |
120.670 |
|
C4 |
C6 |
C5 |
94.893 |
| C4 |
C6 |
H11 |
57.017 |
|
C4 |
C6 |
H12 |
26.003 |
| C5 |
C3 |
H15 |
2.536 |
|
C5 |
C3 |
H16 |
40.528 |
| C5 |
C6 |
H11 |
62.519 |
|
C5 |
C6 |
H12 |
79.577 |
| C6 |
C4 |
H13 |
110.474 |
|
C6 |
C4 |
H14 |
107.569 |
| C6 |
C5 |
H9 |
78.692 |
|
C6 |
C5 |
H10 |
95.028 |
| H9 |
C5 |
H10 |
31.590 |
|
H11 |
C6 |
H12 |
31.025 |
| H13 |
C4 |
H14 |
106.604 |
|
H15 |
C3 |
H16 |
39.285 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.040 |
|
|
|
| 2 |
C |
-0.090 |
|
|
|
| 3 |
C |
-0.172 |
|
|
|
| 4 |
C |
0.034 |
|
|
|
| 5 |
O |
-0.461 |
|
|
|
| 6 |
O |
-0.439 |
|
|
|
| 7 |
H |
0.100 |
|
|
|
| 8 |
H |
0.065 |
|
|
|
| 9 |
H |
0.069 |
|
|
|
| 10 |
H |
0.079 |
|
|
|
| 11 |
H |
0.091 |
|
|
|
| 12 |
H |
0.080 |
|
|
|
| 13 |
H |
0.058 |
|
|
|
| 14 |
H |
0.068 |
|
|
|
| 15 |
H |
0.276 |
|
|
|
| 16 |
H |
0.283 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.293 |
0.774 |
0.138 |
1.513 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.698 |
5.621 |
-3.512 |
| y |
5.621 |
-37.140 |
2.101 |
| z |
-3.512 |
2.101 |
-39.035 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.389 |
5.621 |
-3.512 |
| y |
5.621 |
0.727 |
2.101 |
| z |
-3.512 |
2.101 |
-2.116 |
|
| Polar |
|---|
| 3z2-r2 | -4.232 |
|---|
| x2-y2 | 0.442 |
|---|
| xy | 5.621 |
|---|
| xz | -3.512 |
|---|
| yz | 2.101 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.158 |
0.282 |
-0.145 |
| y |
0.282 |
7.684 |
0.102 |
| z |
-0.145 |
0.102 |
7.191 |
<r2> (average value of r
2) Å
2
| <r2> |
185.174 |
| (<r2>)1/2 |
13.608 |