Jump to
S1C2
Energy calculated at HF/daug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -270.134172 |
| Energy at 298.15K | -270.144922 |
| HF Energy | -270.134172 |
| Nuclear repulsion energy | 237.196750 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/daug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3273 |
2961 |
17.38 |
|
|
|
| 2 |
A' |
3215 |
2908 |
59.40 |
|
|
|
| 3 |
A' |
3180 |
2876 |
14.60 |
|
|
|
| 4 |
A' |
3167 |
2865 |
12.69 |
|
|
|
| 5 |
A' |
3150 |
2849 |
37.91 |
|
|
|
| 6 |
A' |
3142 |
2842 |
13.62 |
|
|
|
| 7 |
A' |
1972 |
1784 |
230.35 |
|
|
|
| 8 |
A' |
1626 |
1471 |
6.12 |
|
|
|
| 9 |
A' |
1608 |
1454 |
1.47 |
|
|
|
| 10 |
A' |
1585 |
1434 |
21.27 |
|
|
|
| 11 |
A' |
1576 |
1425 |
0.70 |
|
|
|
| 12 |
A' |
1546 |
1399 |
30.90 |
|
|
|
| 13 |
A' |
1536 |
1390 |
10.95 |
|
|
|
| 14 |
A' |
1519 |
1374 |
12.76 |
|
|
|
| 15 |
A' |
1444 |
1306 |
6.79 |
|
|
|
| 16 |
A' |
1283 |
1160 |
67.69 |
|
|
|
| 17 |
A' |
1216 |
1100 |
0.25 |
|
|
|
| 18 |
A' |
1110 |
1004 |
0.32 |
|
|
|
| 19 |
A' |
1042 |
943 |
7.02 |
|
|
|
| 20 |
A' |
962 |
871 |
6.35 |
|
|
|
| 21 |
A' |
886 |
802 |
0.04 |
|
|
|
| 22 |
A' |
639 |
578 |
19.88 |
|
|
|
| 23 |
A' |
422 |
382 |
1.45 |
|
|
|
| 24 |
A' |
362 |
327 |
3.28 |
|
|
|
| 25 |
A' |
186 |
168 |
5.41 |
|
|
|
| 26 |
A" |
3224 |
2916 |
97.23 |
|
|
|
| 27 |
A" |
3219 |
2912 |
1.93 |
|
|
|
| 28 |
A" |
3189 |
2885 |
9.56 |
|
|
|
| 29 |
A" |
3164 |
2862 |
8.96 |
|
|
|
| 30 |
A" |
1616 |
1461 |
5.37 |
|
|
|
| 31 |
A" |
1598 |
1446 |
9.48 |
|
|
|
| 32 |
A" |
1436 |
1299 |
0.05 |
|
|
|
| 33 |
A" |
1364 |
1233 |
0.10 |
|
|
|
| 34 |
A" |
1251 |
1131 |
0.86 |
|
|
|
| 35 |
A" |
1064 |
963 |
0.96 |
|
|
|
| 36 |
A" |
904 |
818 |
0.09 |
|
|
|
| 37 |
A" |
783 |
708 |
3.00 |
|
|
|
| 38 |
A" |
520 |
470 |
1.39 |
|
|
|
| 39 |
A" |
262 |
237 |
0.01 |
|
|
|
| 40 |
A" |
112 |
101 |
0.16 |
|
|
|
| 41 |
A" |
105 |
95 |
1.56 |
|
|
|
| 42 |
A" |
16 |
14 |
0.28 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 32735.2 cm
-1
Scaled (by 0.9046) Zero Point Vibrational Energy (zpe) 29612.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/daug-cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.408 |
1.352 |
0.000 |
| C2 |
-1.465 |
0.154 |
0.000 |
| C3 |
0.000 |
0.569 |
0.000 |
| C4 |
0.983 |
-0.584 |
0.000 |
| C5 |
2.449 |
-0.214 |
0.000 |
| O6 |
0.631 |
-1.719 |
0.000 |
| H7 |
-3.442 |
1.031 |
0.000 |
| H8 |
-2.256 |
1.975 |
0.876 |
| H9 |
-2.256 |
1.975 |
-0.876 |
| H10 |
-1.658 |
-0.468 |
0.865 |
| H11 |
-1.658 |
-0.468 |
-0.865 |
| H12 |
0.224 |
1.190 |
-0.865 |
| H13 |
0.224 |
1.190 |
0.865 |
| H14 |
3.054 |
-1.107 |
0.000 |
| H15 |
2.682 |
0.385 |
-0.873 |
| H16 |
2.682 |
0.385 |
0.873 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5249 | 2.5316 | 3.9046 | 5.1026 | 4.3201 | 1.0834 | 1.0852 | 1.0852 | 2.1506 | 2.1506 | 2.7745 | 2.7745 | 5.9897 | 5.2536 | 5.2536 |
C2 | 1.5249 | | 1.5224 | 2.5563 | 3.9306 | 2.8102 | 2.1632 | 2.1698 | 2.1698 | 1.0832 | 1.0832 | 2.1614 | 2.1614 | 4.6911 | 4.2438 | 4.2438 | C3 | 2.5316 | 1.5224 | | 1.5157 | 2.5710 | 2.3734 | 3.4729 | 2.7983 | 2.7983 | 2.1388 | 2.1388 | 1.0878 | 1.0878 | 3.4839 | 2.8265 | 2.8265 | C4 | 3.9046 | 2.5563 | 1.5157 | | 1.5120 | 1.1876 | 4.7105 | 4.2195 | 4.2195 | 2.7815 | 2.7815 | 2.1148 | 2.1148 | 2.1360 | 2.1424 | 2.1424 | C5 | 5.1026 | 3.9306 | 2.5710 | 1.5120 | | 2.3599 | 6.0209 | 5.2621 | 5.2621 | 4.2047 | 4.2047 | 2.7694 | 2.7694 | 1.0791 | 1.0844 | 1.0844 | O6 | 4.3201 | 2.8102 | 2.3734 | 1.1876 | 2.3599 | | 4.9141 | 4.7686 | 4.7686 | 2.7481 | 2.7481 | 3.0616 | 3.0616 | 2.4988 | 3.0653 | 3.0653 | H7 | 1.0834 | 2.1632 | 3.4729 | 4.7105 | 6.0209 | 4.9141 | | 1.7509 | 1.7509 | 2.4857 | 2.4857 | 3.7699 | 3.7699 | 6.8387 | 6.2196 | 6.2196 | H8 | 1.0852 | 2.1698 | 2.7983 | 4.2195 | 5.2621 | 4.7686 | 1.7509 | | 1.7521 | 2.5148 | 3.0589 | 3.1296 | 2.6007 | 6.2014 | 5.4742 | 5.1872 | H9 | 1.0852 | 2.1698 | 2.7983 | 4.2195 | 5.2621 | 4.7686 | 1.7509 | 1.7521 | | 3.0589 | 2.5148 | 2.6007 | 3.1296 | 6.2014 | 5.1872 | 5.4742 | H10 | 2.1506 | 1.0832 | 2.1388 | 2.7815 | 4.2047 | 2.7481 | 2.4857 | 2.5148 | 3.0589 | | 1.7309 | 3.0470 | 2.5080 | 4.8331 | 4.7526 | 4.4231 | H11 | 2.1506 | 1.0832 | 2.1388 | 2.7815 | 4.2047 | 2.7481 | 2.4857 | 3.0589 | 2.5148 | 1.7309 | | 2.5080 | 3.0470 | 4.8331 | 4.4231 | 4.7526 | H12 | 2.7745 | 2.1614 | 1.0878 | 2.1148 | 2.7694 | 3.0616 | 3.7699 | 3.1296 | 2.6007 | 3.0470 | 2.5080 | | 1.7298 | 3.7464 | 2.5866 | 3.1163 | H13 | 2.7745 | 2.1614 | 1.0878 | 2.1148 | 2.7694 | 3.0616 | 3.7699 | 2.6007 | 3.1296 | 2.5080 | 3.0470 | 1.7298 | | 3.7464 | 3.1163 | 2.5866 | H14 | 5.9897 | 4.6911 | 3.4839 | 2.1360 | 1.0791 | 2.4988 | 6.8387 | 6.2014 | 6.2014 | 4.8331 | 4.8331 | 3.7464 | 3.7464 | | 1.7689 | 1.7689 | H15 | 5.2536 | 4.2438 | 2.8265 | 2.1424 | 1.0844 | 3.0653 | 6.2196 | 5.4742 | 5.1872 | 4.7526 | 4.4231 | 2.5866 | 3.1163 | 1.7689 | | 1.7463 | H16 | 5.2536 | 4.2438 | 2.8265 | 2.1424 | 1.0844 | 3.0653 | 6.2196 | 5.1872 | 5.4742 | 4.4231 | 4.7526 | 3.1163 | 2.5866 | 1.7689 | 1.7463 | |
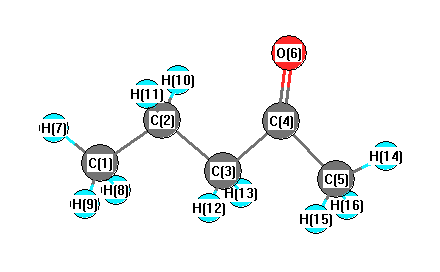 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.357 |
|
C1 |
C2 |
H10 |
109.929 |
| C1 |
C2 |
H11 |
109.929 |
|
C2 |
C1 |
H7 |
110.926 |
| C2 |
C1 |
H8 |
111.355 |
|
C2 |
C1 |
H9 |
111.355 |
| C2 |
C3 |
C4 |
114.587 |
|
C2 |
C3 |
H12 |
110.707 |
| C2 |
C3 |
H13 |
110.707 |
|
C3 |
C2 |
H10 |
109.181 |
| C3 |
C2 |
H11 |
109.181 |
|
C3 |
C4 |
C5 |
116.242 |
| C3 |
C4 |
O6 |
122.334 |
|
C4 |
C3 |
H12 |
107.510 |
| C4 |
C3 |
H13 |
107.510 |
|
C4 |
C5 |
H14 |
109.919 |
| C4 |
C5 |
H15 |
110.116 |
|
C4 |
C5 |
H16 |
110.116 |
| C5 |
C4 |
O6 |
121.424 |
|
H7 |
C1 |
H8 |
107.682 |
| H7 |
C1 |
H9 |
107.682 |
|
H8 |
C1 |
H9 |
107.663 |
| H10 |
C2 |
H11 |
106.064 |
|
H12 |
C3 |
H13 |
105.338 |
| H14 |
C5 |
H15 |
109.694 |
|
H14 |
C5 |
H16 |
109.694 |
| H15 |
C5 |
H16 |
107.262 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/daug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-1.114 |
|
|
|
| 2 |
C |
-0.050 |
|
|
|
| 3 |
C |
-0.447 |
|
|
|
| 4 |
C |
1.523 |
|
|
|
| 5 |
C |
-1.437 |
|
|
|
| 6 |
O |
-1.414 |
|
|
|
| 7 |
H |
0.343 |
|
|
|
| 8 |
H |
0.385 |
|
|
|
| 9 |
H |
0.385 |
|
|
|
| 10 |
H |
0.265 |
|
|
|
| 11 |
H |
0.265 |
|
|
|
| 12 |
H |
0.062 |
|
|
|
| 13 |
H |
0.062 |
|
|
|
| 14 |
H |
0.535 |
|
|
|
| 15 |
H |
0.318 |
|
|
|
| 16 |
H |
0.318 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.759 |
2.912 |
0.000 |
3.009 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.473 |
1.604 |
0.000 |
| y |
1.604 |
-45.145 |
0.000 |
| z |
0.000 |
0.000 |
-37.217 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.708 |
1.604 |
0.000 |
| y |
1.604 |
-8.800 |
0.000 |
| z |
0.000 |
0.000 |
3.092 |
|
| Polar |
|---|
| 3z2-r2 | 6.184 |
|---|
| x2-y2 | 9.673 |
|---|
| xy | 1.604 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.296 |
-0.406 |
0.000 |
| y |
-0.406 |
9.305 |
0.000 |
| z |
0.000 |
0.000 |
7.534 |
<r2> (average value of r
2) Å
2
| <r2> |
227.194 |
| (<r2>)1/2 |
15.073 |
Jump to
S1C1
Energy calculated at HF/daug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -270.134172 |
| Energy at 298.15K | -270.144921 |
| HF Energy | -270.134172 |
| Nuclear repulsion energy | 237.191237 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/daug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3273 |
2961 |
17.40 |
|
|
|
| 2 |
A' |
3215 |
2908 |
59.47 |
|
|
|
| 3 |
A' |
3180 |
2877 |
14.51 |
|
|
|
| 4 |
A' |
3167 |
2865 |
12.77 |
|
|
|
| 5 |
A' |
3150 |
2849 |
37.84 |
|
|
|
| 6 |
A' |
3142 |
2842 |
13.60 |
|
|
|
| 7 |
A' |
1973 |
1784 |
230.42 |
|
|
|
| 8 |
A' |
1626 |
1471 |
6.12 |
|
|
|
| 9 |
A' |
1608 |
1454 |
1.48 |
|
|
|
| 10 |
A' |
1585 |
1434 |
21.26 |
|
|
|
| 11 |
A' |
1576 |
1425 |
0.73 |
|
|
|
| 12 |
A' |
1546 |
1399 |
31.10 |
|
|
|
| 13 |
A' |
1536 |
1390 |
10.77 |
|
|
|
| 14 |
A' |
1519 |
1374 |
12.71 |
|
|
|
| 15 |
A' |
1444 |
1306 |
6.76 |
|
|
|
| 16 |
A' |
1283 |
1160 |
67.73 |
|
|
|
| 17 |
A' |
1216 |
1100 |
0.25 |
|
|
|
| 18 |
A' |
1110 |
1004 |
0.32 |
|
|
|
| 19 |
A' |
1042 |
943 |
7.02 |
|
|
|
| 20 |
A' |
962 |
871 |
6.38 |
|
|
|
| 21 |
A' |
886 |
802 |
0.04 |
|
|
|
| 22 |
A' |
639 |
578 |
19.87 |
|
|
|
| 23 |
A' |
422 |
382 |
1.45 |
|
|
|
| 24 |
A' |
362 |
327 |
3.28 |
|
|
|
| 25 |
A' |
186 |
168 |
5.41 |
|
|
|
| 26 |
A" |
3224 |
2916 |
96.54 |
|
|
|
| 27 |
A" |
3219 |
2912 |
2.48 |
|
|
|
| 28 |
A" |
3189 |
2885 |
9.70 |
|
|
|
| 29 |
A" |
3164 |
2862 |
8.96 |
|
|
|
| 30 |
A" |
1616 |
1461 |
5.37 |
|
|
|
| 31 |
A" |
1598 |
1446 |
9.47 |
|
|
|
| 32 |
A" |
1436 |
1299 |
0.05 |
|
|
|
| 33 |
A" |
1364 |
1233 |
0.10 |
|
|
|
| 34 |
A" |
1251 |
1131 |
0.87 |
|
|
|
| 35 |
A" |
1064 |
963 |
0.96 |
|
|
|
| 36 |
A" |
904 |
818 |
0.08 |
|
|
|
| 37 |
A" |
783 |
708 |
3.00 |
|
|
|
| 38 |
A" |
520 |
470 |
1.39 |
|
|
|
| 39 |
A" |
262 |
237 |
0.01 |
|
|
|
| 40 |
A" |
112 |
101 |
0.15 |
|
|
|
| 41 |
A" |
105 |
95 |
1.56 |
|
|
|
| 42 |
A" |
15 |
14 |
0.28 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 32734.8 cm
-1
Scaled (by 0.9046) Zero Point Vibrational Energy (zpe) 29611.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/daug-cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.407 |
1.353 |
0.000 |
| C2 |
-1.465 |
0.154 |
0.000 |
| C3 |
0.000 |
0.569 |
0.000 |
| C4 |
0.983 |
-0.585 |
0.000 |
| C5 |
2.449 |
-0.214 |
0.000 |
| O6 |
0.631 |
-1.719 |
0.000 |
| H7 |
-3.442 |
1.032 |
0.000 |
| H8 |
-2.255 |
1.975 |
0.876 |
| H9 |
-2.255 |
1.975 |
-0.876 |
| H10 |
-1.659 |
-0.468 |
0.865 |
| H11 |
-1.659 |
-0.468 |
-0.865 |
| H12 |
0.224 |
1.190 |
-0.865 |
| H13 |
0.224 |
1.190 |
0.865 |
| H14 |
3.054 |
-1.108 |
0.000 |
| H15 |
2.682 |
0.385 |
-0.873 |
| H16 |
2.682 |
0.385 |
0.873 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5251 | 2.5317 | 3.9047 | 5.1026 | 4.3207 | 1.0834 | 1.0852 | 1.0852 | 2.1506 | 2.1506 | 2.7743 | 2.7743 | 5.9898 | 5.2536 | 5.2536 |
C2 | 1.5251 | | 1.5223 | 2.5565 | 3.9307 | 2.8108 | 2.1634 | 2.1700 | 2.1700 | 1.0832 | 1.0832 | 2.1613 | 2.1613 | 4.6913 | 4.2440 | 4.2440 | C3 | 2.5317 | 1.5223 | | 1.5155 | 2.5709 | 2.3735 | 3.4730 | 2.7984 | 2.7984 | 2.1389 | 2.1389 | 1.0877 | 1.0877 | 3.4838 | 2.8266 | 2.8266 | C4 | 3.9047 | 2.5565 | 1.5155 | | 1.5121 | 1.1876 | 4.7108 | 4.2196 | 4.2196 | 2.7819 | 2.7819 | 2.1148 | 2.1148 | 2.1361 | 2.1426 | 2.1426 | C5 | 5.1026 | 3.9307 | 2.5709 | 1.5121 | | 2.3598 | 6.0210 | 5.2619 | 5.2619 | 4.2051 | 4.2051 | 2.7693 | 2.7693 | 1.0791 | 1.0844 | 1.0844 | O6 | 4.3207 | 2.8108 | 2.3735 | 1.1876 | 2.3598 | | 4.9150 | 4.7692 | 4.7692 | 2.7489 | 2.7489 | 3.0617 | 3.0617 | 2.4987 | 3.0653 | 3.0653 | H7 | 1.0834 | 2.1634 | 3.4730 | 4.7108 | 6.0210 | 4.9150 | | 1.7508 | 1.7508 | 2.4857 | 2.4857 | 3.7697 | 3.7697 | 6.8391 | 6.2197 | 6.2197 | H8 | 1.0852 | 2.1700 | 2.7984 | 4.2196 | 5.2619 | 4.7692 | 1.7508 | | 1.7521 | 2.5148 | 3.0589 | 3.1294 | 2.6005 | 6.2014 | 5.4741 | 5.1871 | H9 | 1.0852 | 2.1700 | 2.7984 | 4.2196 | 5.2619 | 4.7692 | 1.7508 | 1.7521 | | 3.0589 | 2.5148 | 2.6005 | 3.1294 | 6.2014 | 5.1871 | 5.4741 | H10 | 2.1506 | 1.0832 | 2.1389 | 2.7819 | 4.2051 | 2.7489 | 2.4857 | 2.5148 | 3.0589 | | 1.7309 | 3.0470 | 2.5080 | 4.8337 | 4.7530 | 4.4236 | H11 | 2.1506 | 1.0832 | 2.1389 | 2.7819 | 4.2051 | 2.7489 | 2.4857 | 3.0589 | 2.5148 | 1.7309 | | 2.5080 | 3.0470 | 4.8337 | 4.4236 | 4.7530 | H12 | 2.7743 | 2.1613 | 1.0877 | 2.1148 | 2.7693 | 3.0617 | 3.7697 | 3.1294 | 2.6005 | 3.0470 | 2.5080 | | 1.7298 | 3.7463 | 2.5867 | 3.1164 | H13 | 2.7743 | 2.1613 | 1.0877 | 2.1148 | 2.7693 | 3.0617 | 3.7697 | 2.6005 | 3.1294 | 2.5080 | 3.0470 | 1.7298 | | 3.7463 | 3.1164 | 2.5867 | H14 | 5.9898 | 4.6913 | 3.4838 | 2.1361 | 1.0791 | 2.4987 | 6.8391 | 6.2014 | 6.2014 | 4.8337 | 4.8337 | 3.7463 | 3.7463 | | 1.7688 | 1.7688 | H15 | 5.2536 | 4.2440 | 2.8266 | 2.1426 | 1.0844 | 3.0653 | 6.2197 | 5.4741 | 5.1871 | 4.7530 | 4.4236 | 2.5867 | 3.1164 | 1.7688 | | 1.7463 | H16 | 5.2536 | 4.2440 | 2.8266 | 2.1426 | 1.0844 | 3.0653 | 6.2197 | 5.1871 | 5.4741 | 4.4236 | 4.7530 | 3.1164 | 2.5867 | 1.7688 | 1.7463 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.354 |
|
C1 |
C2 |
H10 |
109.924 |
| C1 |
C2 |
H11 |
109.924 |
|
C2 |
C1 |
H7 |
110.930 |
| C2 |
C1 |
H8 |
111.355 |
|
C2 |
C1 |
H9 |
111.355 |
| C2 |
C3 |
C4 |
114.605 |
|
C2 |
C3 |
H12 |
110.695 |
| C2 |
C3 |
H13 |
110.695 |
|
C3 |
C2 |
H10 |
109.187 |
| C3 |
C2 |
H11 |
109.187 |
|
C3 |
C4 |
C5 |
116.237 |
| C3 |
C4 |
O6 |
122.352 |
|
C4 |
C3 |
H12 |
107.514 |
| C4 |
C3 |
H13 |
107.514 |
|
C4 |
C5 |
H14 |
109.919 |
| C4 |
C5 |
H15 |
110.126 |
|
C4 |
C5 |
H16 |
110.126 |
| C5 |
C4 |
O6 |
121.411 |
|
H7 |
C1 |
H8 |
107.679 |
| H7 |
C1 |
H9 |
107.679 |
|
H8 |
C1 |
H9 |
107.665 |
| H10 |
C2 |
H11 |
106.063 |
|
H12 |
C3 |
H13 |
105.335 |
| H14 |
C5 |
H15 |
109.686 |
|
H14 |
C5 |
H16 |
109.686 |
| H15 |
C5 |
H16 |
107.259 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/daug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-1.114 |
|
|
|
| 2 |
C |
-0.050 |
|
|
|
| 3 |
C |
-0.446 |
|
|
|
| 4 |
C |
1.524 |
|
|
|
| 5 |
C |
-1.437 |
|
|
|
| 6 |
O |
-1.415 |
|
|
|
| 7 |
H |
0.344 |
|
|
|
| 8 |
H |
0.385 |
|
|
|
| 9 |
H |
0.385 |
|
|
|
| 10 |
H |
0.265 |
|
|
|
| 11 |
H |
0.265 |
|
|
|
| 12 |
H |
0.062 |
|
|
|
| 13 |
H |
0.062 |
|
|
|
| 14 |
H |
0.535 |
|
|
|
| 15 |
H |
0.318 |
|
|
|
| 16 |
H |
0.318 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.758 |
2.912 |
0.000 |
3.009 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.474 |
1.603 |
0.000 |
| y |
1.603 |
-45.147 |
0.000 |
| z |
0.000 |
0.000 |
-37.217 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.708 |
1.603 |
0.000 |
| y |
1.603 |
-8.801 |
0.000 |
| z |
0.000 |
0.000 |
3.093 |
|
| Polar |
|---|
| 3z2-r2 | 6.187 |
|---|
| x2-y2 | 9.673 |
|---|
| xy | 1.603 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.296 |
-0.406 |
0.000 |
| y |
-0.406 |
9.306 |
0.000 |
| z |
0.000 |
0.000 |
7.534 |
<r2> (average value of r
2) Å
2
| <r2> |
227.214 |
| (<r2>)1/2 |
15.074 |