Jump to
S1C2
Energy calculated at HF/daug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -192.260864 |
| Energy at 298.15K | -192.265739 |
| HF Energy | -192.260864 |
| Nuclear repulsion energy | 149.424554 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/daug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3386 |
3063 |
0.17 |
|
|
|
| 2 |
A1 |
3365 |
3044 |
22.29 |
|
|
|
| 3 |
A1 |
3348 |
3028 |
4.78 |
|
|
|
| 4 |
A1 |
1563 |
1414 |
16.30 |
|
|
|
| 5 |
A1 |
1503 |
1359 |
32.28 |
|
|
|
| 6 |
A1 |
1196 |
1082 |
2.40 |
|
|
|
| 7 |
A1 |
1094 |
989 |
0.75 |
|
|
|
| 8 |
A1 |
971 |
878 |
0.58 |
|
|
|
| 9 |
A1 |
895 |
809 |
0.23 |
|
|
|
| 10 |
A2 |
962 |
871 |
0.00 |
|
|
|
| 11 |
A2 |
769 |
696 |
0.00 |
|
|
|
| 12 |
A2 |
537 |
485 |
0.00 |
|
|
|
| 13 |
B1 |
968 |
876 |
10.11 |
|
|
|
| 14 |
B1 |
837 |
757 |
1.61 |
|
|
|
| 15 |
B1 |
715 |
647 |
129.38 |
|
|
|
| 16 |
B1 |
543 |
491 |
5.46 |
|
|
|
| 17 |
B2 |
3375 |
3053 |
15.83 |
|
|
|
| 18 |
B2 |
3354 |
3034 |
0.12 |
|
|
|
| 19 |
B2 |
1597 |
1445 |
0.38 |
|
|
|
| 20 |
B2 |
1406 |
1272 |
0.16 |
|
|
|
| 21 |
B2 |
1332 |
1205 |
0.36 |
|
|
|
| 22 |
B2 |
1113 |
1007 |
10.23 |
|
|
|
| 23 |
B2 |
993 |
899 |
0.64 |
|
|
|
| 24 |
B2 |
701 |
634 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18260.6 cm
-1
Scaled (by 0.9046) Zero Point Vibrational Energy (zpe) 16518.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/daug-cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
1.183 |
| C2 |
0.000 |
1.162 |
0.351 |
| C3 |
0.000 |
0.746 |
-0.943 |
| C4 |
0.000 |
-0.746 |
-0.943 |
| C5 |
0.000 |
-1.162 |
0.351 |
| H6 |
0.000 |
0.000 |
2.255 |
| H7 |
0.000 |
2.174 |
0.695 |
| H8 |
0.000 |
1.360 |
-1.820 |
| H9 |
0.000 |
-1.360 |
-1.820 |
| H10 |
0.000 |
-2.174 |
0.695 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.4290 | 2.2531 | 2.2531 | 1.4290 | 1.0714 | 2.2284 | 3.2965 | 3.2965 | 2.2284 |
C2 | 1.4290 | | 1.3591 | 2.3046 | 2.3232 | 2.2301 | 1.0698 | 2.1798 | 3.3270 | 3.3537 | C3 | 2.2531 | 1.3591 | | 1.4911 | 2.3046 | 3.2833 | 2.1739 | 1.0705 | 2.2805 | 3.3481 | C4 | 2.2531 | 2.3046 | 1.4911 | | 1.3591 | 3.2833 | 3.3481 | 2.2805 | 1.0705 | 2.1739 | C5 | 1.4290 | 2.3232 | 2.3046 | 1.3591 | | 2.2301 | 3.3537 | 3.3270 | 2.1798 | 1.0698 | H6 | 1.0714 | 2.2301 | 3.2833 | 3.2833 | 2.2301 | | 2.6757 | 4.2953 | 4.2953 | 2.6757 | H7 | 2.2284 | 1.0698 | 2.1739 | 3.3481 | 3.3537 | 2.6757 | | 2.6439 | 4.3377 | 4.3488 | H8 | 3.2965 | 2.1798 | 1.0705 | 2.2805 | 3.3270 | 4.2953 | 2.6439 | | 2.7193 | 4.3377 | H9 | 3.2965 | 3.3270 | 2.2805 | 1.0705 | 2.1798 | 4.2953 | 4.3377 | 2.7193 | | 2.6439 | H10 | 2.2284 | 3.3537 | 3.3481 | 2.1739 | 1.0698 | 2.6757 | 4.3488 | 4.3377 | 2.6439 | |
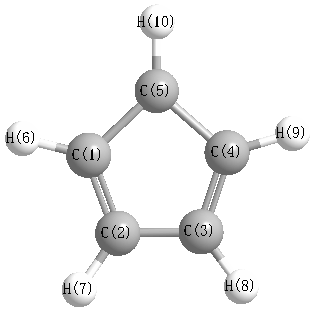 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
107.796 |
|
C1 |
C2 |
H7 |
125.596 |
| C1 |
C5 |
C4 |
107.796 |
|
C1 |
C5 |
H10 |
125.596 |
| C2 |
C1 |
C5 |
108.757 |
|
C2 |
C1 |
H6 |
125.622 |
| C2 |
C3 |
C4 |
107.825 |
|
C2 |
C3 |
H8 |
127.169 |
| C3 |
C2 |
H7 |
126.607 |
|
C3 |
C4 |
C5 |
107.825 |
| C3 |
C4 |
H9 |
125.006 |
|
C4 |
C3 |
H8 |
125.006 |
| C4 |
C5 |
H10 |
126.607 |
|
C5 |
C1 |
H6 |
125.622 |
| C5 |
C4 |
H9 |
127.169 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/daug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.783 |
|
|
|
| 2 |
C |
-0.866 |
|
|
|
| 3 |
C |
-0.799 |
|
|
|
| 4 |
C |
-0.799 |
|
|
|
| 5 |
C |
-0.866 |
|
|
|
| 6 |
H |
0.882 |
|
|
|
| 7 |
H |
0.795 |
|
|
|
| 8 |
H |
0.820 |
|
|
|
| 9 |
H |
0.820 |
|
|
|
| 10 |
H |
0.795 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.153 |
0.153 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.781 |
0.000 |
0.000 |
| y |
0.000 |
-28.083 |
0.000 |
| z |
0.000 |
0.000 |
-25.868 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.805 |
0.000 |
0.000 |
| y |
0.000 |
1.741 |
0.000 |
| z |
0.000 |
0.000 |
5.064 |
|
| Polar |
|---|
| 3z2-r2 | 10.127 |
|---|
| x2-y2 | -5.697 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.659 |
0.000 |
0.000 |
| y |
0.000 |
8.524 |
0.000 |
| z |
0.000 |
0.000 |
9.270 |
<r2> (average value of r
2) Å
2
| <r2> |
87.501 |
| (<r2>)1/2 |
9.354 |
Jump to
S1C1
Energy calculated at HF/daug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -192.259928 |
| Energy at 298.15K | |
| HF Energy | -192.259928 |
| Nuclear repulsion energy | 149.583809 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/daug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3389 |
3066 |
0.00 |
|
|
|
| 2 |
A1 |
3381 |
3058 |
14.16 |
|
|
|
| 3 |
A1 |
3352 |
3032 |
0.75 |
|
|
|
| 4 |
A1 |
1708 |
1546 |
0.19 |
|
|
|
| 5 |
A1 |
1545 |
1398 |
0.52 |
|
|
|
| 6 |
A1 |
1218 |
1102 |
0.35 |
|
|
|
| 7 |
A1 |
1127 |
1020 |
7.84 |
|
|
|
| 8 |
A1 |
1078 |
975 |
3.62 |
|
|
|
| 9 |
A1 |
895 |
810 |
0.36 |
|
|
|
| 10 |
A2 |
1048 |
948 |
0.00 |
|
|
|
| 11 |
A2 |
828 |
749 |
0.00 |
|
|
|
| 12 |
A2 |
563 |
510 |
0.00 |
|
|
|
| 13 |
B1 |
929 |
840 |
14.42 |
|
|
|
| 14 |
B1 |
802 |
726 |
20.13 |
|
|
|
| 15 |
B1 |
715 |
647 |
107.29 |
|
|
|
| 16 |
B1 |
524 |
474 |
7.14 |
|
|
|
| 17 |
B2 |
3365 |
3044 |
34.35 |
|
|
|
| 18 |
B2 |
3352 |
3033 |
8.25 |
|
|
|
| 19 |
B2 |
1501 |
1358 |
44.78 |
|
|
|
| 20 |
B2 |
1407 |
1273 |
0.02 |
|
|
|
| 21 |
B2 |
1265 |
1144 |
11.94 |
|
|
|
| 22 |
B2 |
1012 |
915 |
0.66 |
|
|
|
| 23 |
B2 |
922 |
834 |
0.01 |
|
|
|
| 24 |
B2 |
2226i |
2013i |
1979.34 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16851.2 cm
-1
Scaled (by 0.9046) Zero Point Vibrational Energy (zpe) 15243.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/daug-cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
1.219 |
| C2 |
0.000 |
1.123 |
0.393 |
| C3 |
0.000 |
0.667 |
-1.002 |
| C4 |
0.000 |
-0.667 |
-1.002 |
| C5 |
0.000 |
-1.123 |
0.393 |
| H6 |
0.000 |
0.000 |
2.288 |
| H7 |
0.000 |
2.146 |
0.709 |
| H8 |
0.000 |
1.310 |
-1.858 |
| H9 |
0.000 |
-1.310 |
-1.858 |
| H10 |
0.000 |
-2.146 |
0.709 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.3938 | 2.3196 | 2.3196 | 1.3938 | 1.0689 | 2.2060 | 3.3444 | 3.3444 | 2.2060 |
C2 | 1.3938 | | 1.4683 | 2.2700 | 2.2457 | 2.2024 | 1.0710 | 2.2590 | 3.3149 | 3.2842 | C3 | 2.3196 | 1.4683 | | 1.3347 | 2.2700 | 3.3575 | 2.2620 | 1.0701 | 2.1548 | 3.2933 | C4 | 2.3196 | 2.2700 | 1.3347 | | 1.4683 | 3.3575 | 3.2933 | 2.1548 | 1.0701 | 2.2620 | C5 | 1.3938 | 2.2457 | 2.2700 | 1.4683 | | 2.2024 | 3.2842 | 3.3149 | 2.2590 | 1.0710 | H6 | 1.0689 | 2.2024 | 3.3575 | 3.3575 | 2.2024 | | 2.6644 | 4.3481 | 4.3481 | 2.6644 | H7 | 2.2060 | 1.0710 | 2.2620 | 3.2933 | 3.2842 | 2.6644 | | 2.6996 | 4.3055 | 4.2924 | H8 | 3.3444 | 2.2590 | 1.0701 | 2.1548 | 3.3149 | 4.3481 | 2.6996 | | 2.6207 | 4.3055 | H9 | 3.3444 | 3.3149 | 2.1548 | 1.0701 | 2.2590 | 4.3481 | 4.3055 | 2.6207 | | 2.6996 | H10 | 2.2060 | 3.2842 | 3.2933 | 2.2620 | 1.0710 | 2.6644 | 4.2924 | 4.3055 | 2.6996 | |
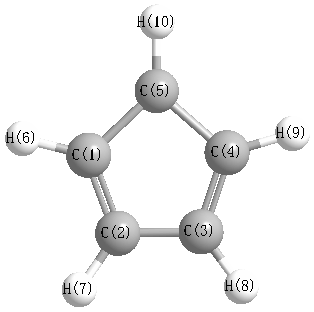 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
108.259 |
|
C1 |
C2 |
H7 |
126.521 |
| C1 |
C5 |
C4 |
108.259 |
|
C1 |
C5 |
H10 |
126.521 |
| C2 |
C1 |
C5 |
107.338 |
|
C2 |
C1 |
H6 |
126.331 |
| C2 |
C3 |
C4 |
108.072 |
|
C2 |
C3 |
H8 |
124.997 |
| C3 |
C2 |
H7 |
125.220 |
|
C3 |
C4 |
C5 |
108.072 |
| C3 |
C4 |
H9 |
126.932 |
|
C4 |
C3 |
H8 |
126.932 |
| C4 |
C5 |
H10 |
125.220 |
|
C5 |
C1 |
H6 |
126.331 |
| C5 |
C4 |
H9 |
124.997 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/daug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.831 |
|
|
|
| 2 |
C |
-0.804 |
|
|
|
| 3 |
C |
-0.830 |
|
|
|
| 4 |
C |
-0.830 |
|
|
|
| 5 |
C |
-0.804 |
|
|
|
| 6 |
H |
0.898 |
|
|
|
| 7 |
H |
0.797 |
|
|
|
| 8 |
H |
0.803 |
|
|
|
| 9 |
H |
0.803 |
|
|
|
| 10 |
H |
0.797 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.027 |
0.027 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.832 |
0.000 |
0.000 |
| y |
0.000 |
-25.795 |
0.000 |
| z |
0.000 |
0.000 |
-28.118 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.876 |
0.000 |
0.000 |
| y |
0.000 |
5.181 |
0.000 |
| z |
0.000 |
0.000 |
1.696 |
|
| Polar |
|---|
| 3z2-r2 | 3.391 |
|---|
| x2-y2 | -8.038 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.689 |
0.000 |
0.000 |
| y |
0.000 |
10.701 |
0.000 |
| z |
0.000 |
0.000 |
8.563 |
<r2> (average value of r
2) Å
2
| <r2> |
87.365 |
| (<r2>)1/2 |
9.347 |