Jump to
S1C2
Energy calculated at HF/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -548.969608 |
| Energy at 298.15K | |
| HF Energy | -548.969608 |
| Nuclear repulsion energy | 357.688850 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/cc-pVDZ
Geometric Data calculated at HF/cc-pVDZ
Point Group is D4h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
1.004 |
0.000 |
| C2 |
1.004 |
0.000 |
0.000 |
| C3 |
0.000 |
-1.004 |
0.000 |
| C4 |
-1.004 |
0.000 |
0.000 |
| F5 |
0.000 |
2.301 |
0.000 |
| F6 |
2.301 |
0.000 |
0.000 |
| F7 |
0.000 |
-2.301 |
0.000 |
| F8 |
-2.301 |
0.000 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.4202 | 2.0084 | 1.4202 | 1.2970 | 2.5108 | 3.3055 | 2.5108 |
C2 | 1.4202 | | 1.4202 | 2.0084 | 2.5108 | 1.2970 | 2.5108 | 3.3055 | C3 | 2.0084 | 1.4202 | | 1.4202 | 3.3055 | 2.5108 | 1.2970 | 2.5108 | C4 | 1.4202 | 2.0084 | 1.4202 | | 2.5108 | 3.3055 | 2.5108 | 1.2970 | F5 | 1.2970 | 2.5108 | 3.3055 | 2.5108 | | 3.2545 | 4.6025 | 3.2545 | F6 | 2.5108 | 1.2970 | 2.5108 | 3.3055 | 3.2545 | | 3.2545 | 4.6025 | F7 | 3.3055 | 2.5108 | 1.2970 | 2.5108 | 4.6025 | 3.2545 | | 3.2545 | F8 | 2.5108 | 3.3055 | 2.5108 | 1.2970 | 3.2545 | 4.6025 | 3.2545 | |
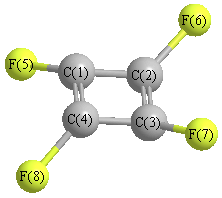 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
135.000 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
135.000 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
135.000 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
135.000 |
| C3 |
C2 |
F6 |
135.000 |
|
C3 |
C4 |
F8 |
135.000 |
| C4 |
C1 |
F5 |
135.000 |
|
C4 |
C3 |
F7 |
135.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.049 |
|
|
|
| 2 |
C |
0.426 |
|
|
|
| 3 |
C |
-0.049 |
|
|
|
| 4 |
C |
0.426 |
|
|
|
| 5 |
F |
-0.224 |
|
|
|
| 6 |
F |
-0.153 |
|
|
|
| 7 |
F |
-0.224 |
|
|
|
| 8 |
F |
-0.153 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.980 |
0.000 |
0.000 |
| y |
0.000 |
-49.882 |
0.000 |
| z |
0.000 |
0.000 |
-40.596 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.260 |
0.000 |
0.000 |
| y |
0.000 |
-10.094 |
0.000 |
| z |
0.000 |
0.000 |
3.834 |
|
| Polar |
|---|
| 3z2-r2 | 7.669 |
|---|
| x2-y2 | 10.902 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.549 |
0.000 |
0.000 |
| y |
0.000 |
6.077 |
0.000 |
| z |
0.000 |
0.000 |
2.900 |
<r2> (average value of r
2) Å
2
| <r2> |
241.803 |
| (<r2>)1/2 |
15.550 |
Jump to
S1C1
Energy calculated at HF/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -549.044719 |
| Energy at 298.15K | -549.045083 |
| HF Energy | -549.044719 |
| Nuclear repulsion energy | 356.241781 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2090 |
1898 |
0.00 |
|
|
|
| 2 |
Ag |
1380 |
1253 |
0.00 |
|
|
|
| 3 |
Ag |
689 |
626 |
0.00 |
|
|
|
| 4 |
Ag |
242 |
219 |
0.00 |
|
|
|
| 5 |
Ag |
180 |
164 |
0.00 |
|
|
|
| 6 |
Au |
1469 |
1333 |
541.64 |
|
|
|
| 7 |
Au |
1021 |
927 |
106.91 |
|
|
|
| 8 |
Au |
751 |
682 |
0.00 |
|
|
|
| 9 |
Au |
243 |
220 |
0.27 |
|
|
|
| 10 |
Au |
182 |
165 |
0.00 |
|
|
|
| 11 |
Bg |
1507 |
1369 |
0.00 |
|
|
|
| 12 |
Bg |
826 |
750 |
0.00 |
|
|
|
| 13 |
Bg |
600 |
545 |
0.00 |
|
|
|
| 14 |
Bg |
512 |
465 |
0.00 |
|
|
|
| 15 |
Bu |
2094 |
1902 |
98.98 |
|
|
|
| 16 |
Bu |
1058 |
961 |
239.03 |
|
|
|
| 17 |
Bu |
329 |
299 |
11.08 |
|
|
|
| 18 |
Bu |
242 |
219 |
1.49 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 7707.1 cm
-1
Scaled (by 0.908) Zero Point Vibrational Energy (zpe) 6998.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/cc-pVDZ
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.785 |
0.652 |
| C2 |
-0.000 |
-0.785 |
0.652 |
| C3 |
-0.000 |
-0.785 |
-0.652 |
| C4 |
0.000 |
0.785 |
-0.652 |
| F5 |
-0.000 |
1.633 |
1.635 |
| F6 |
0.000 |
-1.633 |
1.635 |
| F7 |
0.000 |
-1.633 |
-1.635 |
| F8 |
-0.000 |
1.633 |
-1.635 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.5694 | 2.0402 | 1.3036 | 1.2984 | 2.6100 | 3.3277 | 2.4388 |
C2 | 1.5694 | | 1.3036 | 2.0402 | 2.6100 | 1.2984 | 2.4388 | 3.3277 | C3 | 2.0402 | 1.3036 | | 1.5694 | 3.3277 | 2.4388 | 1.2984 | 2.6100 | C4 | 1.3036 | 2.0402 | 1.5694 | | 2.4388 | 3.3277 | 2.6100 | 1.2984 | F5 | 1.2984 | 2.6100 | 3.3277 | 2.4388 | | 3.2665 | 4.6214 | 3.2691 | F6 | 2.6100 | 1.2984 | 2.4388 | 3.3277 | 3.2665 | | 3.2691 | 4.6214 | F7 | 3.3277 | 2.4388 | 1.2984 | 2.6100 | 4.6214 | 3.2691 | | 3.2665 | F8 | 2.4388 | 3.3277 | 2.6100 | 1.2984 | 3.2691 | 4.6214 | 3.2665 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
130.810 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
139.190 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
130.810 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
139.190 |
| C3 |
C2 |
F6 |
139.190 |
|
C3 |
C4 |
F8 |
130.810 |
| C4 |
C1 |
F5 |
139.190 |
|
C4 |
C3 |
F7 |
130.810 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.196 |
|
|
|
| 2 |
C |
0.196 |
|
|
|
| 3 |
C |
0.196 |
|
|
|
| 4 |
C |
0.196 |
|
|
|
| 5 |
F |
-0.196 |
|
|
|
| 6 |
F |
-0.196 |
|
|
|
| 7 |
F |
-0.196 |
|
|
|
| 8 |
F |
-0.196 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.150 |
0.000 |
0.000 |
| y |
0.000 |
-44.673 |
0.000 |
| z |
0.000 |
0.000 |
-44.325 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.348 |
0.000 |
0.000 |
| y |
0.000 |
-2.435 |
0.000 |
| z |
0.000 |
0.000 |
-1.913 |
|
| Polar |
|---|
| 3z2-r2 | -3.826 |
|---|
| x2-y2 | 4.522 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.840 |
0.000 |
0.000 |
| y |
0.000 |
4.998 |
0.000 |
| z |
0.000 |
0.000 |
6.887 |
<r2> (average value of r
2) Å
2
| <r2> |
244.075 |
| (<r2>)1/2 |
15.623 |