Jump to
S1C2
Energy calculated at HF/6-311G*
| | hartrees |
|---|
| Energy at 0K | -577.212866 |
| Energy at 298.15K | -577.220728 |
| Nuclear repulsion energy | 158.609627 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3260 |
2948 |
46.13 |
|
|
|
| 2 |
A' |
3245 |
2935 |
47.59 |
|
|
|
| 3 |
A' |
3205 |
2899 |
14.60 |
|
|
|
| 4 |
A' |
3178 |
2874 |
37.46 |
|
|
|
| 5 |
A' |
1642 |
1485 |
5.61 |
|
|
|
| 6 |
A' |
1628 |
1472 |
0.37 |
|
|
|
| 7 |
A' |
1625 |
1469 |
2.59 |
|
|
|
| 8 |
A' |
1553 |
1404 |
0.42 |
|
|
|
| 9 |
A' |
1515 |
1370 |
16.74 |
|
|
|
| 10 |
A' |
1416 |
1281 |
36.55 |
|
|
|
| 11 |
A' |
1208 |
1093 |
1.13 |
|
|
|
| 12 |
A' |
1106 |
1001 |
1.68 |
|
|
|
| 13 |
A' |
969 |
876 |
9.86 |
|
|
|
| 14 |
A' |
784 |
709 |
63.84 |
|
|
|
| 15 |
A' |
386 |
349 |
4.71 |
|
|
|
| 16 |
A' |
254 |
230 |
2.52 |
|
|
|
| 17 |
A" |
3319 |
3001 |
30.94 |
|
|
|
| 18 |
A" |
3248 |
2938 |
63.71 |
|
|
|
| 19 |
A" |
3222 |
2914 |
7.48 |
|
|
|
| 20 |
A" |
1633 |
1477 |
8.04 |
|
|
|
| 21 |
A" |
1442 |
1305 |
0.17 |
|
|
|
| 22 |
A" |
1363 |
1233 |
0.84 |
|
|
|
| 23 |
A" |
1205 |
1089 |
1.60 |
|
|
|
| 24 |
A" |
942 |
852 |
0.00 |
|
|
|
| 25 |
A" |
805 |
728 |
2.40 |
|
|
|
| 26 |
A" |
254 |
230 |
0.04 |
|
|
|
| 27 |
A" |
127 |
115 |
1.92 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22266.8 cm
-1
Scaled (by 0.9044) Zero Point Vibrational Energy (zpe) 20138.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.588 |
0.000 |
| C2 |
0.904 |
-0.632 |
0.000 |
| C3 |
2.378 |
-0.231 |
0.000 |
| Cl4 |
-1.743 |
0.125 |
0.000 |
| H5 |
0.146 |
1.198 |
0.878 |
| H6 |
0.146 |
1.198 |
-0.878 |
| H7 |
0.682 |
-1.239 |
-0.871 |
| H8 |
0.682 |
-1.239 |
0.871 |
| H9 |
3.013 |
-1.109 |
0.000 |
| H10 |
2.632 |
0.357 |
-0.877 |
| H11 |
2.632 |
0.357 |
0.877 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5181 | 2.5153 | 1.8033 | 1.0789 | 1.0789 | 2.1364 | 2.1364 | 3.4586 | 2.7840 | 2.7840 |
C2 | 1.5181 | | 1.5282 | 2.7521 | 2.1659 | 2.1659 | 1.0846 | 1.0846 | 2.1630 | 2.1757 | 2.1757 | C3 | 2.5153 | 1.5282 | | 4.1361 | 2.7918 | 2.7918 | 2.1566 | 2.1566 | 1.0842 | 1.0857 | 1.0857 | Cl4 | 1.8033 | 2.7521 | 4.1361 | | 2.3430 | 2.3430 | 2.9154 | 2.9154 | 4.9133 | 4.4677 | 4.4677 | H5 | 1.0789 | 2.1659 | 2.7918 | 2.3430 | | 1.7568 | 3.0472 | 2.4951 | 3.7834 | 3.1574 | 2.6244 | H6 | 1.0789 | 2.1659 | 2.7918 | 2.3430 | 1.7568 | | 2.4951 | 3.0472 | 3.7834 | 2.6244 | 3.1574 | H7 | 2.1364 | 1.0846 | 2.1566 | 2.9154 | 3.0472 | 2.4951 | | 1.7415 | 2.4914 | 2.5195 | 3.0664 | H8 | 2.1364 | 1.0846 | 2.1566 | 2.9154 | 2.4951 | 3.0472 | 1.7415 | | 2.4914 | 3.0664 | 2.5195 | H9 | 3.4586 | 2.1630 | 1.0842 | 4.9133 | 3.7834 | 3.7834 | 2.4914 | 2.4914 | | 1.7505 | 1.7505 | H10 | 2.7840 | 2.1757 | 1.0857 | 4.4677 | 3.1574 | 2.6244 | 2.5195 | 3.0664 | 1.7505 | | 1.7542 | H11 | 2.7840 | 2.1757 | 1.0857 | 4.4677 | 2.6244 | 3.1574 | 3.0664 | 2.5195 | 1.7505 | 1.7542 | |
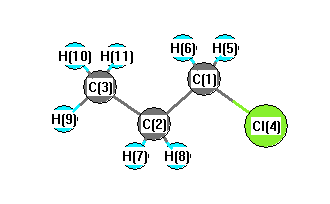 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
111.315 |
|
C1 |
C2 |
H7 |
109.202 |
| C1 |
C2 |
H8 |
109.202 |
|
C2 |
C1 |
Cl4 |
111.624 |
| C2 |
C1 |
H5 |
111.912 |
|
C2 |
C1 |
H6 |
111.912 |
| C2 |
C3 |
H9 |
110.639 |
|
C2 |
C3 |
H10 |
111.562 |
| C2 |
C3 |
H11 |
111.562 |
|
C3 |
C2 |
H7 |
110.100 |
| C3 |
C2 |
H8 |
110.100 |
|
Cl4 |
C1 |
H5 |
106.018 |
| Cl4 |
C1 |
H6 |
106.018 |
|
H5 |
C1 |
H6 |
109.016 |
| H7 |
C2 |
H8 |
106.808 |
|
H9 |
C3 |
H10 |
107.555 |
| H9 |
C3 |
H11 |
107.555 |
|
H10 |
C3 |
H11 |
107.778 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.426 |
|
|
|
| 2 |
C |
-0.408 |
|
|
|
| 3 |
C |
-0.622 |
|
|
|
| 4 |
Cl |
-0.151 |
|
|
|
| 5 |
H |
0.255 |
|
|
|
| 6 |
H |
0.255 |
|
|
|
| 7 |
H |
0.226 |
|
|
|
| 8 |
H |
0.226 |
|
|
|
| 9 |
H |
0.223 |
|
|
|
| 10 |
H |
0.210 |
|
|
|
| 11 |
H |
0.210 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.580 |
0.334 |
0.000 |
2.602 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.080 |
0.162 |
0.000 |
| y |
0.162 |
-33.086 |
0.000 |
| z |
0.000 |
0.000 |
-33.229 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.922 |
0.162 |
0.000 |
| y |
0.162 |
2.068 |
0.000 |
| z |
0.000 |
0.000 |
1.854 |
|
| Polar |
|---|
| 3z2-r2 | 3.709 |
|---|
| x2-y2 | -3.993 |
|---|
| xy | 0.162 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.350 |
0.162 |
0.000 |
| y |
0.162 |
5.457 |
0.000 |
| z |
0.000 |
0.000 |
5.123 |
<r2> (average value of r
2) Å
2
| <r2> |
152.971 |
| (<r2>)1/2 |
12.368 |
Jump to
S1C1
Energy calculated at HF/6-311G*
| | hartrees |
|---|
| Energy at 0K | -577.212371 |
| Energy at 298.15K | -577.220338 |
| Nuclear repulsion energy | 162.308444 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3320 |
3003 |
22.82 |
|
|
|
| 2 |
A |
3265 |
2953 |
79.09 |
|
|
|
| 3 |
A |
3261 |
2949 |
13.25 |
|
|
|
| 4 |
A |
3241 |
2932 |
60.42 |
|
|
|
| 5 |
A |
3220 |
2912 |
7.34 |
|
|
|
| 6 |
A |
3184 |
2880 |
26.04 |
|
|
|
| 7 |
A |
3174 |
2871 |
43.39 |
|
|
|
| 8 |
A |
1640 |
1483 |
5.07 |
|
|
|
| 9 |
A |
1636 |
1480 |
6.37 |
|
|
|
| 10 |
A |
1620 |
1465 |
5.42 |
|
|
|
| 11 |
A |
1614 |
1459 |
2.04 |
|
|
|
| 12 |
A |
1559 |
1410 |
4.16 |
|
|
|
| 13 |
A |
1514 |
1369 |
0.84 |
|
|
|
| 14 |
A |
1469 |
1329 |
51.57 |
|
|
|
| 15 |
A |
1406 |
1272 |
3.24 |
|
|
|
| 16 |
A |
1350 |
1221 |
1.34 |
|
|
|
| 17 |
A |
1208 |
1092 |
0.50 |
|
|
|
| 18 |
A |
1183 |
1070 |
1.20 |
|
|
|
| 19 |
A |
1122 |
1014 |
3.94 |
|
|
|
| 20 |
A |
973 |
880 |
4.16 |
|
|
|
| 21 |
A |
920 |
832 |
8.55 |
|
|
|
| 22 |
A |
857 |
775 |
18.37 |
|
|
|
| 23 |
A |
696 |
630 |
40.21 |
|
|
|
| 24 |
A |
451 |
408 |
2.64 |
|
|
|
| 25 |
A |
317 |
286 |
0.70 |
|
|
|
| 26 |
A |
231 |
209 |
0.88 |
|
|
|
| 27 |
A |
140 |
127 |
1.07 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22285.8 cm
-1
Scaled (by 0.9044) Zero Point Vibrational Energy (zpe) 20155.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.182 |
0.872 |
0.315 |
| C2 |
-1.141 |
0.558 |
-0.363 |
| C3 |
-1.826 |
-0.706 |
0.145 |
| Cl4 |
1.460 |
-0.346 |
-0.068 |
| H5 |
0.579 |
1.820 |
-0.013 |
| H6 |
0.093 |
0.880 |
1.390 |
| H7 |
-1.787 |
1.416 |
-0.191 |
| H8 |
-0.985 |
0.498 |
-1.435 |
| H9 |
-2.781 |
-0.846 |
-0.349 |
| H10 |
-1.224 |
-1.588 |
-0.040 |
| H11 |
-2.013 |
-0.649 |
1.214 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5191 | 2.5596 | 1.8065 | 1.0780 | 1.0791 | 2.1051 | 2.1366 | 3.4887 | 2.8556 | 2.8181 |
C2 | 1.5191 | | 1.5257 | 2.7693 | 2.1608 | 2.1678 | 1.0882 | 1.0848 | 2.1593 | 2.1718 | 2.1693 | C3 | 2.5596 | 1.5257 | | 3.3127 | 3.4908 | 2.7831 | 2.1495 | 2.1574 | 1.0845 | 1.0829 | 1.0861 | Cl4 | 1.8065 | 2.7693 | 3.3127 | | 2.3386 | 2.3447 | 3.6971 | 2.9260 | 4.2797 | 2.9582 | 3.7149 | H5 | 1.0780 | 2.1608 | 3.4908 | 2.3386 | | 1.7571 | 2.4068 | 2.4933 | 4.3015 | 3.8550 | 3.7836 | H6 | 1.0791 | 2.1678 | 2.7831 | 2.3447 | 1.7571 | | 2.5148 | 3.0481 | 3.7763 | 3.1414 | 2.6085 | H7 | 2.1051 | 1.0882 | 2.1495 | 3.6971 | 2.4068 | 2.5148 | | 1.7422 | 2.4757 | 3.0600 | 2.5080 | H8 | 2.1366 | 1.0848 | 2.1574 | 2.9260 | 2.4933 | 3.0481 | 1.7422 | | 2.4917 | 2.5203 | 3.0639 | H9 | 3.4887 | 2.1593 | 1.0845 | 4.2797 | 4.3015 | 3.7763 | 2.4757 | 2.4917 | | 1.7515 | 1.7521 | H10 | 2.8556 | 2.1718 | 1.0829 | 2.9582 | 3.8550 | 3.1414 | 3.0600 | 2.5203 | 1.7515 | | 1.7537 | H11 | 2.8181 | 2.1693 | 1.0861 | 3.7149 | 3.7836 | 2.6085 | 2.5080 | 3.0639 | 1.7521 | 1.7537 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
114.419 |
|
C1 |
C2 |
H7 |
106.511 |
| C1 |
C2 |
H8 |
109.142 |
|
C2 |
C1 |
Cl4 |
112.477 |
| C2 |
C1 |
H5 |
111.477 |
|
C2 |
C1 |
H6 |
111.978 |
| C2 |
C3 |
H9 |
110.497 |
|
C2 |
C3 |
H10 |
111.600 |
| C2 |
C3 |
H11 |
111.208 |
|
C3 |
C2 |
H7 |
109.503 |
| C3 |
C2 |
H8 |
110.326 |
|
Cl4 |
C1 |
H5 |
105.543 |
| Cl4 |
C1 |
H6 |
105.924 |
|
H5 |
C1 |
H6 |
109.083 |
| H7 |
C2 |
H8 |
106.590 |
|
H9 |
C3 |
H10 |
107.825 |
| H9 |
C3 |
H11 |
107.648 |
|
H10 |
C3 |
H11 |
107.901 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.430 |
|
|
|
| 2 |
C |
-0.410 |
|
|
|
| 3 |
C |
-0.614 |
|
|
|
| 4 |
Cl |
-0.156 |
|
|
|
| 5 |
H |
0.261 |
|
|
|
| 6 |
H |
0.254 |
|
|
|
| 7 |
H |
0.212 |
|
|
|
| 8 |
H |
0.227 |
|
|
|
| 9 |
H |
0.215 |
|
|
|
| 10 |
H |
0.238 |
|
|
|
| 11 |
H |
0.203 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-2.011 |
1.484 |
0.360 |
2.525 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.111 |
0.395 |
0.185 |
| y |
0.395 |
-32.423 |
0.519 |
| z |
0.185 |
0.519 |
-33.310 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.245 |
0.395 |
0.185 |
| y |
0.395 |
2.288 |
0.519 |
| z |
0.185 |
0.519 |
0.957 |
|
| Polar |
|---|
| 3z2-r2 | 1.913 |
|---|
| x2-y2 | -3.688 |
|---|
| xy | 0.395 |
|---|
| xz | 0.185 |
|---|
| yz | 0.519 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.246 |
-0.745 |
-0.179 |
| y |
-0.745 |
6.218 |
0.187 |
| z |
-0.179 |
0.187 |
5.287 |
<r2> (average value of r
2) Å
2
| <r2> |
131.927 |
| (<r2>)1/2 |
11.486 |