Jump to
S1C2
S1C3
Energy calculated at HF/6-31G
| | hartrees |
|---|
| Energy at 0K | -580.009155 |
| Energy at 298.15K | |
| HF Energy | -580.009155 |
| Nuclear repulsion energy | 78.432123 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2330 |
2104 |
0.00 |
425.94 |
0.22 |
0.36 |
| 2 |
Ag |
986 |
891 |
0.00 |
28.05 |
0.53 |
0.69 |
| 3 |
Ag |
622 |
561 |
0.00 |
143.01 |
0.28 |
0.43 |
| 4 |
Au |
586 |
529 |
0.00 |
0.00 |
0.00 |
0.00 |
| 5 |
B1u |
2318 |
2093 |
131.16 |
0.00 |
0.00 |
0.00 |
| 6 |
B1u |
895 |
808 |
106.73 |
0.00 |
0.00 |
0.00 |
| 7 |
B2g |
173i |
156i |
0.00 |
137.59 |
0.75 |
0.86 |
| 8 |
B2u |
2356 |
2127 |
266.21 |
0.00 |
0.00 |
0.00 |
| 9 |
B2u |
396 |
357 |
29.23 |
0.00 |
0.00 |
0.00 |
| 10 |
B3g |
2345 |
2117 |
0.00 |
237.82 |
0.75 |
0.86 |
| 11 |
B3g |
624 |
564 |
0.00 |
7.88 |
0.75 |
0.86 |
| 12 |
B3u |
589 |
532 |
5.92 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6936.8 cm
-1
Scaled (by 0.9029) Zero Point Vibrational Energy (zpe) 6263.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-31G
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
1.076 |
| Si2 |
0.000 |
0.000 |
-1.076 |
| H3 |
0.000 |
1.257 |
1.859 |
| H4 |
0.000 |
-1.257 |
1.859 |
| H5 |
0.000 |
1.257 |
-1.859 |
| H6 |
0.000 |
-1.257 |
-1.859 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1520 | 1.4807 | 1.4807 | 3.1927 | 3.1927 |
Si2 | 2.1520 | | 3.1927 | 3.1927 | 1.4807 | 1.4807 | H3 | 1.4807 | 3.1927 | | 2.5137 | 3.7177 | 4.4878 | H4 | 1.4807 | 3.1927 | 2.5137 | | 4.4878 | 3.7177 | H5 | 3.1927 | 1.4807 | 3.7177 | 4.4878 | | 2.5137 | H6 | 3.1927 | 1.4807 | 4.4878 | 3.7177 | 2.5137 | |
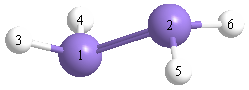 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
121.917 |
|
Si1 |
Si2 |
H6 |
121.917 |
| Si2 |
Si1 |
H3 |
121.917 |
|
Si2 |
Si1 |
H4 |
121.917 |
| H3 |
Si1 |
H4 |
116.166 |
|
H5 |
Si2 |
H6 |
116.166 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.275 |
|
|
|
| 2 |
Si |
0.275 |
|
|
|
| 3 |
H |
-0.138 |
|
|
|
| 4 |
H |
-0.138 |
|
|
|
| 5 |
H |
-0.138 |
|
|
|
| 6 |
H |
-0.138 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.857 |
0.000 |
0.000 |
| y |
0.000 |
-30.699 |
0.000 |
| z |
0.000 |
0.000 |
-29.588 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.714 |
0.000 |
0.000 |
| y |
0.000 |
0.523 |
0.000 |
| z |
0.000 |
0.000 |
2.191 |
|
| Polar |
|---|
| 3z2-r2 | 4.381 |
|---|
| x2-y2 | -2.158 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.270 |
0.000 |
0.000 |
| y |
0.000 |
6.072 |
0.000 |
| z |
0.000 |
0.000 |
15.492 |
<r2> (average value of r
2) Å
2
| <r2> |
71.951 |
| (<r2>)1/2 |
8.482 |
Jump to
S1C1
S1C3
Energy calculated at HF/6-31G
| | hartrees |
|---|
| Energy at 0K | -580.009452 |
| Energy at 298.15K | |
| HF Energy | -580.009452 |
| Nuclear repulsion energy | 78.011412 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2313 |
2088 |
0.00 |
425.00 |
0.20 |
0.33 |
| 2 |
Ag |
991 |
895 |
0.00 |
44.28 |
0.49 |
0.66 |
| 3 |
Ag |
608 |
549 |
0.00 |
119.65 |
0.33 |
0.50 |
| 4 |
Ag |
234 |
211 |
0.00 |
112.59 |
0.52 |
0.68 |
| 5 |
Au |
2337 |
2110 |
292.67 |
0.00 |
0.00 |
0.00 |
| 6 |
Au |
586 |
529 |
0.35 |
0.00 |
0.33 |
0.50 |
| 7 |
Au |
386 |
349 |
31.01 |
0.00 |
0.00 |
0.00 |
| 8 |
Bg |
2325 |
2099 |
0.00 |
270.65 |
0.75 |
0.86 |
| 9 |
Bg |
633 |
571 |
0.00 |
9.22 |
0.75 |
0.86 |
| 10 |
Bu |
2302 |
2079 |
169.48 |
0.00 |
0.00 |
0.00 |
| 11 |
Bu |
924 |
834 |
168.39 |
0.00 |
0.00 |
0.00 |
| 12 |
Bu |
550 |
497 |
41.36 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 7094.7 cm
-1
Scaled (by 0.9029) Zero Point Vibrational Energy (zpe) 6405.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-31G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.085 |
0.000 |
| Si2 |
0.000 |
-1.085 |
0.000 |
| H3 |
0.294 |
1.838 |
1.245 |
| H4 |
0.294 |
1.838 |
-1.245 |
| H5 |
-0.294 |
-1.838 |
1.245 |
| H6 |
-0.294 |
-1.838 |
-1.245 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1694 | 1.4842 | 1.4842 | 3.1902 | 3.1902 |
Si2 | 2.1694 | | 3.1902 | 3.1902 | 1.4842 | 1.4842 | H3 | 1.4842 | 3.1902 | | 2.4890 | 3.7227 | 4.4782 | H4 | 1.4842 | 3.1902 | 2.4890 | | 4.4782 | 3.7227 | H5 | 3.1902 | 1.4842 | 3.7227 | 4.4782 | | 2.4890 | H6 | 3.1902 | 1.4842 | 4.4782 | 3.7227 | 2.4890 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
120.501 |
|
Si1 |
Si2 |
H6 |
120.501 |
| Si2 |
Si1 |
H3 |
120.501 |
|
Si2 |
Si1 |
H4 |
120.501 |
| H3 |
Si1 |
H4 |
113.970 |
|
H5 |
Si2 |
H6 |
113.970 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.287 |
|
|
|
| 2 |
Si |
0.287 |
|
|
|
| 3 |
H |
-0.144 |
|
|
|
| 4 |
H |
-0.144 |
|
|
|
| 5 |
H |
-0.144 |
|
|
|
| 6 |
H |
-0.144 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.838 |
-0.307 |
0.000 |
| y |
-0.307 |
-29.506 |
0.000 |
| z |
0.000 |
0.000 |
-30.874 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.648 |
-0.307 |
0.000 |
| y |
-0.307 |
2.350 |
0.000 |
| z |
0.000 |
0.000 |
0.298 |
|
| Polar |
|---|
| 3z2-r2 | 0.596 |
|---|
| x2-y2 | -3.332 |
|---|
| xy | -0.307 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.353 |
-1.008 |
0.000 |
| y |
-1.008 |
15.713 |
0.000 |
| z |
0.000 |
0.000 |
6.396 |
<r2> (average value of r
2) Å
2
| <r2> |
72.406 |
| (<r2>)1/2 |
8.509 |
Jump to
S1C1
S1C2
Energy calculated at HF/6-31G
| | hartrees |
|---|
| Energy at 0K | -579.965561 |
| Energy at 298.15K | |
| HF Energy | -579.965561 |
| Nuclear repulsion energy | 69.725896 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2105 |
1900 |
678.62 |
383.36 |
0.33 |
0.50 |
| 2 |
A1 |
1578 |
1425 |
0.13 |
84.69 |
0.17 |
0.29 |
| 3 |
A1 |
1031 |
931 |
103.62 |
123.06 |
0.42 |
0.59 |
| 4 |
A1 |
444 |
401 |
0.13 |
12.19 |
0.35 |
0.52 |
| 5 |
A1 |
372 |
336 |
1.32 |
41.36 |
0.30 |
0.46 |
| 6 |
A2 |
929 |
839 |
0.00 |
2.89 |
0.75 |
0.86 |
| 7 |
A2 |
656 |
592 |
0.00 |
3.24 |
0.75 |
0.86 |
| 8 |
B1 |
1138 |
1028 |
230.88 |
0.85 |
0.75 |
0.86 |
| 9 |
B1 |
989 |
893 |
20.76 |
34.23 |
0.75 |
0.86 |
| 10 |
B2 |
2076 |
1875 |
6.59 |
11.61 |
0.75 |
0.86 |
| 11 |
B2 |
1300 |
1174 |
2941.76 |
4.25 |
0.75 |
0.86 |
| 12 |
B2 |
811 |
732 |
127.29 |
35.79 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 6713.8 cm
-1
Scaled (by 0.9029) Zero Point Vibrational Energy (zpe) 6061.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-31G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.398 |
-0.095 |
| Si2 |
0.000 |
-1.398 |
-0.095 |
| H3 |
-1.034 |
0.000 |
-0.108 |
| H4 |
1.034 |
0.000 |
-0.108 |
| H5 |
0.000 |
1.455 |
1.435 |
| H6 |
0.000 |
-1.455 |
1.435 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.7959 | 1.7392 | 1.7392 | 1.5308 | 3.2375 |
Si2 | 2.7959 | | 1.7392 | 1.7392 | 3.2375 | 1.5308 | H3 | 1.7392 | 1.7392 | | 2.0690 | 2.3595 | 2.3595 | H4 | 1.7392 | 1.7392 | 2.0690 | | 2.3595 | 2.3595 | H5 | 1.5308 | 3.2375 | 2.3595 | 2.3595 | | 2.9105 | H6 | 3.2375 | 1.5308 | 2.3595 | 2.3595 | 2.9105 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
28.198 |
|
Si1 |
Si2 |
H6 |
92.145 |
| Si2 |
Si1 |
H3 |
36.503 |
|
Si2 |
Si1 |
H4 |
36.503 |
| H3 |
Si1 |
H4 |
73.000 |
|
H5 |
Si2 |
H6 |
63.947 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.580 |
|
|
|
| 2 |
Si |
0.580 |
|
|
|
| 3 |
H |
-0.379 |
|
|
|
| 4 |
H |
-0.379 |
|
|
|
| 5 |
H |
-0.201 |
|
|
|
| 6 |
H |
-0.201 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.085 |
1.085 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.373 |
0.000 |
0.000 |
| y |
0.000 |
-32.514 |
0.000 |
| z |
0.000 |
0.000 |
-35.794 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.781 |
0.000 |
0.000 |
| y |
0.000 |
0.069 |
0.000 |
| z |
0.000 |
0.000 |
-4.850 |
|
| Polar |
|---|
| 3z2-r2 | -9.700 |
|---|
| x2-y2 | 3.142 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.984 |
0.000 |
0.000 |
| y |
0.000 |
13.384 |
0.000 |
| z |
0.000 |
0.000 |
7.446 |
<r2> (average value of r
2) Å
2
| <r2> |
85.826 |
| (<r2>)1/2 |
9.264 |